Арнольда киари 1 степени мрт

Аномалия Арнольда-Киари I — опущение миндалин мозжечка в большое затылочное отверстие со сдавливанием продолговатого мозга. Может сочетаться с сирингомиелией, базилярной импрессией или инвагинацией, ассимиляцией атланта. Симптомы коррелируют от степени снижения. Методом выбора в диагностике является магнитно-резонансная томография (МРТ). Оперативное лечение, ламинэктомия (удаление дужек верхних шейных позвонков) с декомпрессивной краниоэктомией задней черепной ямки и пластикой твёрдой мозговой оболочки, применяется только при наличии у пациента неврологического дефицита с отсутствием эффекта от консервативной терапии в течение 2-3 месяцев.
авторы Dr Henry Knipe and Dr Frank Gaillard et al.
Эпидемиология
Аномалия Арнольда-Киари I встречается чаще у женщин [2].
Клиническая картина
В отличие от пороков развития Киари II, III и IV, аномалия Арнольда Киари I часто остается бессимптомной.
Вероятность стать клинических проявлений пропорциональна степени опускания миндалин. Все пациенты с пролабированием миндалин больше 12 мм имеют какую-либо симптоматику, тогда как примерно в 30% пролабирование в диапазоне между 5 и 10 мм протекают бессимптомными [1].
Компрессия ствола мозга (продолговатого мозга) может вызывать сирингомиелию с соответствующими симптомами и клинической картиной (затылочные боли, нарушение глотания, атаксия) разной выраженности, симптомами поражения спинного мозга и др.
Сопутствующие заболевания
Сирингомиелия шейного отдела позвоночника встречается в ~35% (варьирует от 20 до 56%), гидроцефалиия в 30% [1,3] случаев, в обоих случаях считается данные изменения развиваются в результате нарушения ликвородинамики, центральном канале и вокруг спинного мозга.
В ~35% (23-45%) выявляются скелетные аномалии [1, 3]:
- платибазия / базилярная импрессия
- атланто-затылочная ассимиляция
- деформация Шпренгеля (Sprengel)
- синдром Клиппель-Фейля (Klippel-Feil)
Патология
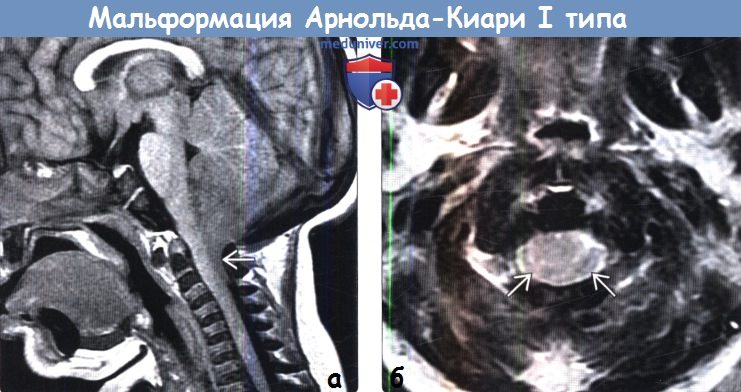
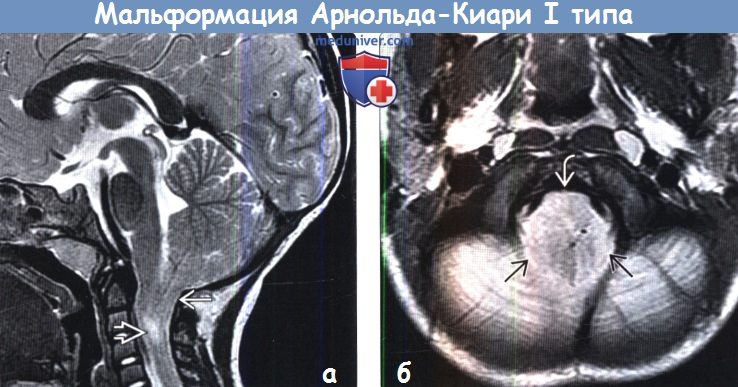
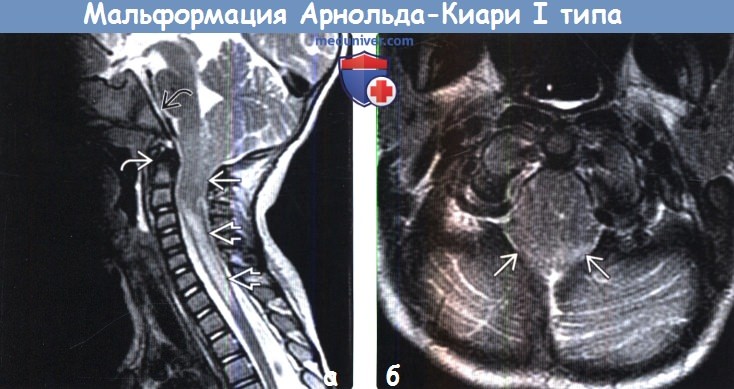
Аномалия Арнольда Киари I характеризуется пролабированием миндалин мозжечка через большое затылочное отверстие, в основном в результате несоответствия между размерами мозжечка и задней черепной ямке. Аномалию Арнольда Киари I следует отличать от эктопии миндалин, которая протекает бессимптомно и является случайной находкой, при которой, миндалины выступают через затылочное отверстие не более чем на 3-5 мм [1-2].
Рентгенологические особенности
Патология выявляется путем измерения максимального расстояния на которое миндалины выступают ниже плоскости большого затылочного отверстия (условной линии между ophisthion и basion), значения используемые для постановки диагноза отличаются у разных авторов [2]:
- выше затылочного отверстия: норма
- <3 мм: норма, может быть использован термин эктопия миндалин
- от 3 до 6 мм: неопределенные данные, необходимо сопоставить с симптоматикой, наличием сирингомиелии и т.д.
- > 6 мм: аномалия Арнольда Киари 1
Некоторые авторы используют более простую градацию [1]:
- выше затылочного отверстия: норма
- <5 мм: норма, может быть использован термин эктопия миндалин
- > 5 мм: аномалия Арнольда Киари 1
Положение миндалин мозжечка меняется с возрастом. У новорожденных миндалины расположены чуть ниже большого затылочного отверстия и спускаются ниже с ростом ребенка, достигая своей низшей точки в возрасте 5 — 15 лет. В дальнейшем они поднимаются на уровень большого затылочного отверстия [3]. Таким образом, снижение миндалин на 5 мм у ребенка будет скорее всего нормой, а взрослом возрасте данные изменения следует рассматривать с подозрением [3].
КТ
Современное объемное сканирование с высоким качеством сагиттальной реформации относительно хорошо визуализирует затылочное отверстие и миндалины, хотя отсутствие контрастности (по сравнению с МРТ) с трудом позволяет провести точную оценку. Чаще патология может быть заподозрена на аксиальных изображениях, когда мозговое вещество охватывает миндалины а спинномозговая жидкость представлена в малом количестве или отсутствует. Данное состояние называется, “переполнением“ затылочного отверстия.
МРТ
МРТ исследование является методом выбора. Сагиттальные срезы наиболее оптимальны для оценки аномалии Арнольда Киари I. Осевые изображения так же дают картину «переполненого» затылочного отверстия.
Лечение и прогноз
Аномалии Арнольда Киари I можно разделить на три этапа (хотя мало данное деление практически не используется в повседневной практике):
- бессимптомная
- компрессия ствола мозга
- сирингомиелия
Оперативное лечение применяется только при наличии у пациента неврологического дефицита с отсутствием эффекта от консервативной терапии в течение 2-3 месяцев. Оно состоит в ламинэктомии (удаление дужек верхних шейных позвонков) с декомпрессивной краниоэктомией задней черепной ямки и пластикой твёрдой мозговой оболочки.
История
Впервые была описана в 1891 году Хансом Киари, Австрийским патологоанатом (1851-1916).
Дифференциальная диагностика
дифференциальный ряд включает в себя:
- эктопия миндалин: <5 мм
- аномалия/порок Арнольда Киари 1.5 (син. бульбарный варинат Киари 1, когда-то считалась вариантом Киари I [4])
- аномалия Арнольда Киари II
- приобретенная эктопия миндалин
- люмбальная пункция
- базилярная инвагинация
Источник
Диагностика мальформации Арнольда-Киари I типа по МРТ, КТа) Терминология: б) Визуализация: 1. Общие характеристики мальформации Арнольда-Киари I типа: 2. КТ при мальформации Арнольда-Киари I типа: 3. МРТ при мальформации Арнольда-Киари I типа: 4. Рекомендации по визуализации мальформации Арнольда-Киари I типа:
в) Дифференциальная диагностика мальформации Арнольда-Киари I типа: 1. Смещение миндалин мозжечка ниже большого затылочного отверстия как вариант нормы: 2. Внутричерепная гипотензия: 3. Приобретенная тонзиллярная грыжа (приобретенная мальформация Арнольда-Киари I типа): 4. Комплексная мальформация Арнольда-Киари:
г) Патология мальформации Арнольда-Киари I типа: 1. Общие характеристики: 2. Стадирование и классификация: 3. Макроскопические и хирургические особенности: 4. Микроскопия:
г) Клиническая картина мальформации Арнольда-Киари I типа: 1. Проявления: 2. Демография: 3. Течение и прогноз: д) Диагностическая памятка: е) Список литературы:
— Также рекомендуем «Мальформация Арнольда-Киари второго типа (МАК II) на МРТ» Редактор: Искандер Милевски. Дата публикации: 24.2.2019 |
Источник

Аномалия Арнольда-Киари — это отклонение в развитии. И конкретно отклонение касается связки головного и спинного мозга. Головной мозг переходит в спинной на уровне большого затылочного отверстия. Четкой границы перехода нет.
Однако расположение ствола мозга относительно костей черепа и шейного отдела позвоночника по разным причинам может меняться. Это несовпадение приводит к сжатию спинного мозга в районе шейного отдела. Нормальная циркуляция спинномозговой жидкости нарушается.
Эта ситуация еще называется мальформация. Название происходит от латинских malus (что означает плохой) и formation (образование). Проще говоря возникает аномалия развития, за которой стоят изменение строения и функций.
В данном случае имеет место мальформация Арнольда-Киари в месте перехода черепа в позвоночник, то есть мальформация головного мозга.
Описали эту патологию австрийский патологоанатом Ханс Киари (в 1891 году) и немецкий патологоанатом Юлиус Арнольд (в 1894 году). Отсюда и сложное название.
Статистика указывает на то, что частота заболевания не такая уж и редкая — до 8.4 заболевших на 100 тысяч населения. Дополнительно к аномалии (до 80% случаев) выявляется сирингомиелия (образование кист в ткани спинного мозга).
Аномалия Арнольда-Киари — что это
Справочно. Аномалия Арнольда-Киари — это нарушение развития, которое приводит к опущению мозжечковых миндалин ниже анатомического уровня и к ущемлению продолговатого мозга.
Нормой считается расположение миндалин мозжечка выше большого затылочного отверстия. В процессе мальформации они могут сместиться даже до уровня второго шейного позвонка. При таком смещении усиливается блокировка тока спинномозговой жидкости.
Не всегда удается распознать это заболевание сразу, а затем происходит его резкая манифестация. Проявление патологии происходит к 25 — 40 годам.
При обнаружении характерных для аномалии симптомов необходимо обратиться к врачу, в противном случае риск развития инфаркта спинного мозга значительно возрастает.
Поскольку речь идет об отклонении от нормального развития организма, заболевание часто называют мальформация Арнольда-Киари.
Особенности мальформации
Справочно. Задний отдел мозга перемещается к большому затылочному отверстию (БЗО), когда у черепной ямки оказываются слишком малые размеры. В итоге формируется порок, возникают нарушения циркуляции ликвора.
Кости черепа не позволяют БЗО менять свой диаметр, поэтому при любых смещениях мозговых структур ущемляются близлежащие ткани. Последствия такого явления могут носить для пациента фатальный характер.
Продолговатый мозг отвечает за работу сердечно-сосудистой и дыхательной системы организма. Его сдавливание приводит не только к неврологическому дефициту, но может давать более опасные последствия, вплоть до смерти больного.
Смещение правого и левого полушарий мозжечка останавливает движение ликвора, что провоцирует гидроцефалию. Водянка повышает риск осложнений тех расстройств, которые уже имеются у пациента.
Процент пациентов с врожденной формой аномалии Киари невелик.
Справочно. Последние данные говорят о приобретенном характере. Дистопия миндалин мозжечка обусловлена быстрым ростом тканей при медленно протекающих изменениях в строении черепа. В медицине также известна под названием эктопия миндалин мозжечка.
Синдром Арнольда-Киари без проявления симптоматики может случайно обнаружиться во время проведения МРТ.
Причины развития аномалии
Мнения медиков о причинах появления аномалии расходятся. Есть несколько теорий, объясняющих то, каким образом развивается порок.
Неврологи выделяют две патологии, приводящие к формированию мальформации Киари (Chiari malformation):
- Развитие плода в утробе матери может пойти с нарушением — черепная ямка окажется меньше анатомической нормы, отделы мозга приобретут обычные параметры.
- Размеры отделов увеличены, при этом параметры задней черепной ямки и БЗО отвечают нормам. Увеличивающийся мозг устремляется к отверстию.
Врожденная аномалия у ребенка развивается, если беременная женщина не контролирует прием лекарств, употребляет алкоголь, курит на ранних сроках беременности.
Кроме того, вирусные инфекции у будущей матери (краснуха, цитомегаловирус) могут пагубно сказаться на развитии плода.
Справочно. Возникновению заболевания могут предшествовать различные родовые травмы, гидроцефалия, сильные повреждения головы у взрослого человека.
Типы аномалии
Рассматриваются четыре типа. Классификация производится с ориентацией на определенные основания.
Существенными признаками оказываются следующие изменения: те, что произошли в головном мозге на структурном уровне, те, что говорят о недоразвитости черепной коробки.
Аномалия Арнольда-Киари 1 типа отличается сдвигом мозжечковых миндалин, сопровождается нарушением циркуляции ликвора.
Последний заполняет узкий канал спинного мозга, вызывая гидромиелию. Указанный тип аномалии носит благоприятный прогноз. Он часто диагностируется у подростковой и взрослой групп населения.
Аномалия Арнольда-Киари 2 типа проявляет себя у новорожденных детей. Здесь наблюдается еще большее смещение отделов. Помимо мальформации, у грудничков диагностируется спинномозговая грыжа, обнаруживается аномальное развитие позвоночного столба.
В области затылка происходит выпячивание мозгового вещества через мягкую оболочку, мозжечок оказывается там же. Такова картина аномалии Арнольда-Киари 3 типа.
Внимание. Аномалия Арнольда-Киари 4 типа дает о себе знать тем, что мозжечок новорожденного оказывается недоразвитым, не занимает должного анатомического положения. Такая патология делает младенца неприспособленным к жизни, летальный исход неизбежен.
Степени тяжести
Сколько живут с аномалией Арнольда-Киари 1-й степени? Такой вопрос часто задают люди, услышавшие свой диагноз. Такая степень тяжести является самой невысокой, клинические проявления могут не отмечаться.
Спровоцировать возникновение симптомов могут черепно-мозговые травмы и повреждения позвоночника в верхней его части. Также запустить процесс может развившаяся нейроинфекция.
Аномалии 2 и 3-й степени уже сопровождаются патологическими изменениями в нервной ткани. У больного часто обнаруживают:
- смещение мозгового вещества;
- кисты проводящих ликвор путей;
- недоразвитость некоторых извилин мозга;
- гипоплазию подкорковых узлов.
Аномалия Арнольда-Киари — симптомы
Говоря о симптомах аномалии, надо, прежде всего, различать вариационные ее типы. Первый тип мальформации сопровождается несколькими синдромами, среди которых: гипертензионный, церебеллярный, сирингомиелический, бульбарный и т.д.
Гипертензионный синдром представляет собой повышение давления внутри черепа (ВЧД). Характерными симптомами будут интенсивные затылочные боли, тошнота, рвота, ригидность шейных мышц.
Церебеллярный синдром характеризуется речевыми расстройствами, нарушениями двигательной функции. При этом отсутствует четкость движений, затруднена мелкая моторика.
Сирингомиелический синдром проявляется потерей чувствительности в конечностях. Больной может получить случайный ожог, не заметив этого сразу. При обследовании обнаруживаются кисты спинного мозга.
Другие типы аномалии сопровождаются более тяжелой симптоматикой.
Справочно. У новорожденных страдает дыхание, возможна его остановка, отмечаются нарушения в глотании. Ребенок не может полноценно питаться. Посинение кожных покровов, гипертонус мышц, нистагм — вот основные проявления аномалии этих типов.
Возможные осложнения
Мальформация в некоторых случаях провоцирует достаточно опасные осложнения и может привести многих пациентов к инвалидности. Часто отмечаются увеличение ВЧД, дыхательные расстройства, апноэ, на фоне мальформации развиваются инфекционные заболевания легких и мочеполовой системы.
Справочно. Тяжело протекающая патология может стать причиной наступления комы, остановки в работе сердца, в итоге, быстрой смерти.
В запущенных случаях реанимация позволяет лишь поддерживать жизненно важные функции, сдавленный мозг восстановить практически невозможно.
Постановка диагноза
Осмотр невролога, сбор анамнеза представляют собой часть диагностики — они необходимы, но недостаточны.
Энцефалограмма, диагностика нарушения кровообращения в головном мозге и шейном отделе позвоночника могут косвенно показать наличие ВЧД.
С помощью рентгенографии, компьютерной томографии можно зафиксировать дефекты формирования черепной коробки. Но для определения состояния нервной ткани такие способы окажутся малоинформативными.
Справочно. МРТ сегодня считается единственно достоверным методом, позволяющим провести точную диагностику и своевременно распознать синдром Арнольда-Киари.
Процедура предполагает полное обездвиживание пациента, чего легко добиться от взрослого человека. Трудности возникают, когда пациентами являются маленькие дети. В этом случае необходимо применение общего наркоза.
Варианты лечения
После постановки диагноза лечение больного осуществляет нейрохирург или невролог. В исключительных случаях устранение аномалии возможно лишь путем проведения операции.
Если единственный симптом болезни — головная боль, то врачи ограничиваются медикаментозной терапией. Специалисты подбирают препараты:
- устраняющие воспаление (Найз, Ибупрофен, Диклофенак);
- анальгетики (Кеторол);
- спазмолитики (Мидокалм).
Справочно. Показанием к операции является сильное сдавливание отделов мозга, явно выраженные неврологические расстройства, если положительный эффект от приема лекарственных препаратов не наблюдается.
Оперативное вмешательство
Благодаря хирургическому вмешательству можно устранить чрезмерное давление на нервную ткань и привести в норму движение ликвора. Одной из проводимых операций является краниовертебральная декомпрессия, направленная на увеличение размеров задней черепной ямки.
Внимание. Декомпрессия относится к классу травматичных и рискованных операций. Согласно статистике, она приводит к осложнениям у каждого десятого пациента.
Риск летального исхода у прооперированных больных выше, чем у тех, кто операцию не переносил. Нейрохирурги стараются проводить такой тип вмешательства только в самых крайних случаях, когда налицо явные признаки сдавливания мозга.
Иной вариант устранения последствий мальформации предполагает шунтирование, которое способно обеспечить отвод ликвора из черепной коробки. Благодаря имплантации специальных трубок жидкость перетекает в грудную и брюшную полости, ВЧД снижается.
В наиболее острых случаях требуется немедленная госпитализация больного и проведение всего спектра терапевтических, профилактических и коррекционных процедур.
Прогноз выживаемости
Продолжительность жизни зависит от типа аномалии и степени ее тяжести. Первый тип позволяет сделать благоприятный прогноз, поскольку симптоматика либо вовсе отсутствует, либо возникает после получения травм головы.
Если никаких проявлений болезни нет, то продолжительность жизни больных такая же, как и у здоровых людей.
Для пациентов со вторым типом аномалии прогноз хуже, она переносится тяжелее.
Иногда борьба с очаговой неврологической симптоматикой не приносит плодов даже при активном лечении медикаментами. В таком случае требуется хирургическое вмешательство, чтобы впоследствии изменения по неврологической части были менее выраженными.
Справочно. Третий и четвертый типы мальформации являются самыми тяжелыми для пациентов, прогноз зачастую неблагоприятен.
Заболевание затрагивает важные структуры мозга, у больного отмечаются пороки внутренних органов. Часто функции ствола мозга страдают настолько, что нарушения оказываются несовместимыми с жизнью.
Источник