Арнольда киари синдром на мрт

МРТ краниовертебрального перехода позволяет безошибочно диагностировать достаточно редкий порок развития — синдром Арнольда — Киари (альтернативное название — мальформации Киари, аномалия Арнольда — Киари, болезнь Киари). В медицине стандартной классификации этого заболевания нет по сегодняшний день. Общепринятым пониманием является определение, что мальформации Арнольда -Киари — это патологические изменения в зоне задней черепной ямки и костных структур краниовертебрального перехода. В результате данной аномалии миндалины мозжечка спускаются в большое затылочное отверстие, сдавливают спинной мозг, нарушают ликвороциркуляцию, приводя к развитию сирингомиелии. Считается, что это врожденная патология, однако болезнь может манифестировать после 40 лет.
БЕСПЛАТНАЯ
КОНСУЛЬТАЦИЯ О ДИАГНОСТИКЕ
Если сомневаетесь, запишитесь на бесплатную консультацию.
Или проконсультируйтесь по телефону
+7 (812) 209-00-79
Перезвоните мне
Мальформации Киари в зависимости от выраженности подразделяются на 4 типа. В большинстве ситуаций неврологам приходится иметь дело с 1 и 2 типом синдрома Арнольда — Киари.
- Мальформация Киари 1 типа — это первичное смещение или аномальное расположение мозжечка, при котором происходит эффект вклинивания миндалин ниже большого затылочного отверстия.
- Сидром Арнольда-Киари 2 типа — это дислокация цервико-медуллярного сочленения, моста, четвертого желудочка и продолговатого мозга.
- Аномалия Арнольда-Киари 3 типа — это сползание структур задней черепной ямки с проникновением мозжечковых структур в шейный канал.
- Мальформация Киари 4 типа — это аномальное недоразвитие мозжечка без проявления смещения.
Признаки аномалии Арнольда- Киари на МРТ
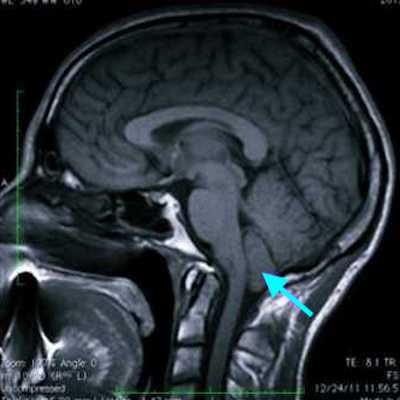 Первичным шагом в обследовании больных с синдромом Арнольда — Киари является прием невролога, который дает направление на МРТ головного мозга и МРТ краниовертебрального перехода. Признаками синдрома Арнольда Киари на МРТ снимках будет:
Первичным шагом в обследовании больных с синдромом Арнольда — Киари является прием невролога, который дает направление на МРТ головного мозга и МРТ краниовертебрального перехода. Признаками синдрома Арнольда Киари на МРТ снимках будет:
- заметное вклинивание миндалин мозжечка ниже большого затылочного отверстия на 5 мм и более;
- вентральная компрессия ствола мозга;
- сирингомиелия;
- наличие признаков внутричерепной гипертензии в виде нарушенной проходимости в путях циркуляции жидкости;
- нарастание ликвородинамических нарушений.
Симптомы
При аномалии Арнольда-Киари пациенты обращаются к невропатологу в основном с тремя жалобами:
- на кашлевые головные боли в затылочной области
- головокружения
- боли в шее.
Однако, в зависимости от стадии развития, другие клинические проявления данного заболевания крайне полиморфны. Недуг может манифестировать себя:
- шаткостью при ходьбе;
- затруднением при глотании;
- обмороками;
- ощущением давления в ушах;
- проблемами со зрительными функциями;
- онемением лица;
- тошнотой, рвотными позывами;
- ошибочным произнесением звуков;
- постепенно нарастающей атрофией в мышцах рук;
- бессонницей.
Дифференциация диагностика аномалии Арнольда-Киари представляет определенную сложность. Она локализуется в узкой зоне организма, где проходят проводящие пути, кровеносные сосуды и ликвороциркуляция, поэтому может давать очень разнообразную симптоматику. Поставить точный диагноз без МРТ исследования головного мозга и краниовертебрального перехода невозможно. Сделать МРТ при подозрении на синдром Арнольда Киари нужно на высокопольном томографе мощностью не менее 1,5 Тесла. В ходе томографии врачи могут отличить данное заболевание от схожих по проявлением:
- опухолей области краниовертебрального перехода;
- наследственной атаксии;
- сосудистых патологий;
- демиелинизирующих заболеваний.
Результаты магнито-резонансного обследования потребуются для проведения оперативного лечения. По данным томографии нейрохирург сможет выстроить план операции по декомпрессии задней черепной ямки с коагуляцией миндалин мозжечка и пластикой твердой мозговой оболочки.
СИРИНГОМИЕЛИЯ НА МРТ
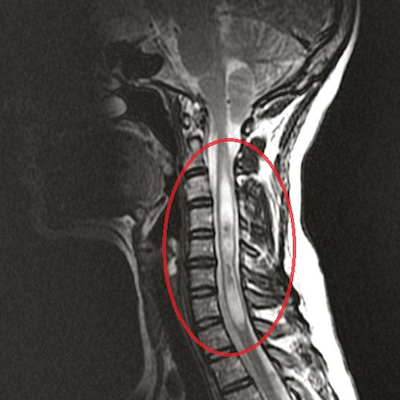 Сирингомиелия, хроническое прогрессирующее заболевание с образованием продольных полостей в спинном мозге, характеризуется распространенностью от 7 до 130 случаев на 100 ООО населения. Это патологический процесс, когда в спинном мозге или продолговатой части головного мозга образуются кисты, сдавливают его и нарушают неврологическую чувствительность. Сирингомиелия может возникать как спорадически, так и быть наследственно-обусловленной. Данная болезнь носит хронический характер и является следствием избыточного роста глиальной ткани. Патологически разросшиеся клетки умирают, и в сером веществе возникают пустоты. Клетки начинают пропускать мозговую жидкость, и она начинает накапливаться, образуя кисту. Киста от продолжающегося притока жидкости продолжает увеличиваться и начинает сдавливать нервные проводящие пути, в первую очередь нарушая проводники болевой и температурной чувствительности. Из-за этого человек при сирингомиелии перестает чувствовать боль и различать холод или тепло руками.
Сирингомиелия, хроническое прогрессирующее заболевание с образованием продольных полостей в спинном мозге, характеризуется распространенностью от 7 до 130 случаев на 100 ООО населения. Это патологический процесс, когда в спинном мозге или продолговатой части головного мозга образуются кисты, сдавливают его и нарушают неврологическую чувствительность. Сирингомиелия может возникать как спорадически, так и быть наследственно-обусловленной. Данная болезнь носит хронический характер и является следствием избыточного роста глиальной ткани. Патологически разросшиеся клетки умирают, и в сером веществе возникают пустоты. Клетки начинают пропускать мозговую жидкость, и она начинает накапливаться, образуя кисту. Киста от продолжающегося притока жидкости продолжает увеличиваться и начинает сдавливать нервные проводящие пути, в первую очередь нарушая проводники болевой и температурной чувствительности. Из-за этого человек при сирингомиелии перестает чувствовать боль и различать холод или тепло руками.
Нередко сирингомиелия сопровождается возникновением патологий в краниовертебральном переходе, в частности Арнольда — Киари, и воспалительные процессы в спинно-мозговых оболочках (Благодаря MPT-исследованиям стало известно, что в основе большинства случаев спорадической сирингомиелии лежат ликвородинамические нарушения, обусловленные мальформацией Арнольда-Киари I типа). Диагностика этого недуга осуществляется неврологом на основании сбора анамнеза и аппаратном обследовании. На сегодняшний день оптимальным методом диагностики сирингомиелии, позволяющим визуализировать морфологическую основу заболевания, является магнитно-резонансная томография. Для того, чтобы понять причину сирингомиелии, врачу нужно сделать МРТ краниовертебрального перехода и МРТ спинного мозга.
Данное заболевание может носить семейно-наследственный характер. Такая форма наблюдается чаще всего у женщин, формируется в среднем возрасте и характеризуется образованием полостей в шейно-грудном регионе и различной степенью изменений области кранио-вертебрального региона.
Семейно-наследственная сирингомиелия представлена двумя нейровизуальными вариантами: ассоциированным (у 90% больных) и неассоциированным с мальформацией Арнольда-Киари I типа (10% больных). У членов семей больных семейно-наследственной сирингомиелией широко распространены невральные и костные аномалии кранио-вертебрального региона типа:
- синдром Арнольда-Киари I типа;
- базилярная инвагинация.
- гипоплазия задней черепной ямки из-за недоразвития костных структур ямки.
В случае подозрения на наследственную форму, врачи рекомендую проводить МРТ обследований головного мозга и шейного отдела у всех членов семьи.
Как проходит томография
В медицинских центрах магнито-резонансное исследование осуществляют по предварительной записи. Сделать это можно бесплатно через Городскую службу записи на МРТ и КТ. Особой подготовки данное обследование не требует. В МРТ кабинет лучше всего прийти за 10-15 минут до назначенного часа, чтобы оформить все документы. Одеться лучше всего в удобную для лежания одежду, на которой не должно быть металлических элементов. В ходе сканирования пациента попросят лечь на томографический стол, на зону скрининга наденут специальную МРТ катушку, передвинут стол внутрь томографа и попросят не двигаться на протяжении диагностики.
Сеанс сканирования в зависимости от модели аппарата МРТ может длиться от 15 до 20 минут. Сама процедура сопровождается достаточно шумной работой установки. Если эти звуки мешают пациенту предлагают надеть специальные наушники для подавления шума. По окончании исследования в большинстве МРТ центров СПб клиент, подождав 30-40 минут, получает МРТ снимки, записанные на электронный носитель, экспертное заключение врача и рекомендации по дальнейшим действиям.
Где сделать МРТ головного мозга и краниовертебрального перехода — адреса клиник
Список первоисточников
- Парамонов Л.В. Клиника, диагностика и хирургическое лечение врождённых аномалий краниовертебрального перехода и шейного отдела позвоночника: автореф. дис. . канд. мед. наук/ Л.В. Парамонов. М., 1969. -26 с.
- Кирьяков В.А. Клиника и диагностика краниовертебральных аномалий.// Журн. невропатол. и психиатрии. 1980. — №11. — С. 1647-1652.
- Косинская Н.С. Краниовертебральные деформации различного происхождения и их влияние на трудоспособность. Методические рекомендации / Н.С. Косинская, Ю.Н. Задворнов, З.К. Быстрова. Л. 1972. -39 с.
- Акберов Р. Ф., Михайлов М. К., Хабибуллин Р. И, Либерман А. В. Комплексная клинико-рентгенологическая диагностика аномалий развития краниовертебральной зоны и позвоночника у детей, подростков и взрослых // Вертеброневрология. -1999. -№ 1-2.-С. 65-72.
- Ахадов Т. А., Белов С. А., Кравцов А. К., Панов В. О. MP-томография в диагностике сирингомиелии // II Междунар. конгресс вертеброневрологов: сборник научных статей. Казань, 1992. — С. 14.
- Валиуллин М. А. Сирингомиелии и мальформация Арнольда-Киари: начальные клинические проявления и результаты хирургического лечения: Автореф. дис. канд. мед. наук. Иркутск, 1996. -23 с.
- Даркшевич JI. О. Сирингомиелия // Курс нервных болезней. Казань: Изд-во братьев Башмаковых, 1909. — Т. 2, вып. 1 и 2. — С. 284-314.
- Завалишин И. А. К диагностике сирингомиелии // Клиническая медицина. -1974. Т. 2, №9. — С. 106 — 110.
- Крупина Н. Е. Особенности наследования мальформации Киари // вестник УрГМА 1999 — №8. — С. 58-61.
- Макаров А. Ю., Холин А. В., Крицкая Л. А. Метод магнитно-резонансной томографии в диагностике сирингомиелии // Вопросы нейрохирургии им. Н. Н. Бурденко. 1991. — №6. — С. 806-810.
- Михайлов М. К., Акберов Р. Ф., Хабибуллин И. Р. Комплексная клинико-рентгенологическая диагностика аномалий развития краниовертебральной зоны и позвоночника у детей, подростков и взрослых: Учеб. Пособие. Казань, 1992 — 58 с.
- Менделевич Е. Г., Чувашаев И. Р. МРТ-морфометрические признаки нарушения циркуляции ликвора у больных сирингомиелией. // Каз. Мед. Журнал -2000-№5-С. 379-381
- Парамонов Л. В. Аномалия Арнольда-Киари // Сов. Медицина. 1967 — №10. -С. 63-67.
- Bogdanov E. I., Ibatullin M. M., Mendelevich E. G. Spontaneous drainage in syringomyelia: MRI findings. // Neuroradiology V. 42 №9 — 2000 — P.676-678
- Taglialatela G, Galasso L. A challenging diagnosis of late-onset tumefactive multiple sclerosis associated to cervicodorsal syringomyelia: doubtful CT, MRI, and bioptic findings: Case report and literature review. Medicine (Baltimore). 2016;95(36):e4585.
Полезная информация

Что покажет МРТ головного мозга
МРТ головного мозга – это хорошо апробированный и надежный метод диагностики, который основывается на свойствах магнитного поля и отличается повышенной информативностью и точностью. Суть метода заключается в том, что под воздействием радиомагнитных импульсов в органах человека возникают ответные волновые сигналы, которые с помощью компьютера томографа преобразовываются в изображения в нескольких
читать далее +

Что покажет МРТ шейного отдела позвоночника
Одним из самых информативных, результативных, неинвазивных методов обнаружения заболеваний позвоночника, эффективность которого проверена десятилетиями, является МРТ шейного отдела позвоночника (или МРТ шеи). В Санкт-Петербурге успешное использование магнитно-резонансной томографии насчитывает сорокалетнюю историю. Данный вид диагностики особенно привлекателен для врачей тем, что безвредное и безболезненное
читать далее +

Что покажет МРТ позвоночника
Заболевания опорно-двигательного аппарата – бич жителей больших мегаполисов, в борьбе с которым достойное место занимает такая диагностическая процедура, как МРТ позвоночника. Магнитно-резонансная томография — это проверенный и хорошо зарекомендовавший себя диагностический метод, отличительными чертами которого является его высокая информативность и неинвазивность. Если перед Вами стоит вопрос, где можно сделать МРТ
читать далее +
Источник
Определение
Аномалия Киари — врожденное смещение структур задней черепной ямки в каудальном направлении.Основную роль сыграл Chiari, который в 1891 г. описал аномалии заднего мозга и дал их классификацию. Арнольд (немецкий анатом,1894 г.) обнаружил сочетание смещения заднего мозга в позвоночный канал и spina bifida и предложил термин мальформация Арнольда-Киари. Сейчас описывают 3 типа этой аномалии.
Морфология
Затылочное смещение миндалин мозжечка, для постановки диагноза, должна быть не менее 4-5мм, а смещенные миндалины имеют треугольную форму. При этом ствол мозга смещается вперед, большая затылочная цистерна полностью или субтотально занята мозжечком. Следует отметить что в возрасте от 5 до 15 лет низкое расположение миндалин является не патологией, а возрастной нормой и требует наблюдения в динамике. Иногда обнаруживается дополнительный выступ на задней поверхности спинного мозга, называемый “шип“ спинного мозга и широкое межталамическое сращение.
Клинические симптомы
Клинические симптомы могут быть переменным. Затылочные головные боли являются одними из самых распространенных жалоб. Кроме того, могут присутствовать головокружение и приступы нистагма. Орофарингеальная дисфункция была описана у маленьких детей.
Так как данная аномалия часто ассоциированна с сингогидроммиелией и довольно часто присутствуюе, поэтому позвоночник должен
также всегда исследоваться.
МРТ и КТ
При МР-томографии первичный диагностический критерий порока развития Арнольда-Киари является тонлиальная эктопия в пределах отверстия. Физиологически, кончик мозжечковых миндалин не должен лежать ниже 5мм линии большого затылочного отверстия у маленьких детей и взрослых, и на 6 мм ниже этой линии у детей в возрасте от 5 до 15 лет (Mikulis et al., 1992).
Кроме того, исследования потока спинномозговой жидкости с закрытым фазовым контрастым измерением могут быть полезным.
Самый важный дифференциальный диагноз для пороки развития Киари I — с внутричерепной гипотензией от хронической утечки СМЖ.

Рис.1
Аномалия Киари I типа (аномалия Арнольда-Киари)

Рис.2
Киари I типа (Арнольда-Киари)
Миндалины мозжечка (tonsilla cerebelli) расположены ниже нижнего края большого затылочного отверстия (БЗО) более, чем на 7мм (среднее значение 13 мм, вариации от 5 до 29 мм) и удлинены (peg-like — вешалка, крючок, колышек).
Часто сочетается с гидроцефалией и сиринго-гидромиелией (30 %), с краниовертебральными аномалиями, такими как остатки
проатала, медианными основными инвагинациями и ассимиляцией C1, смещением ствола мозга кпереди, что приводит к симптому «ступеньки» в месте перехода продолговатого мозга в спинной. При аномалии Киари 1 типа размер задней ямки часто мал.
Клиника связана с симптомами компрессии ствола на уровне БЗО, поражение каудальной группы нервов и мозжечка. Клинические симптомы: головная или цервикальная боль, слабость в конечностях, потеря температурной или болевой чувствительности, диплопия, дисфазия, рвота, дизартрия, мозжечковая атаксия. У части пациентов клинические проявления отсутствуют.
Необходимо помнить, что у детей от 5 до 15 лет смещение на 5 мм не должно рассматриваться как патология. Оперативное лечение состоит в декомпрессии ЗЧЯ путем субокципитальной краниоэктомии и иногда ламинэктомии С1-С3 (у пациентов с клинической симптоматикой).
Аномалия Киари II типа
Через БЗО в большую цистерну мозга пролабируют миндалины мозжечка, червь мозжечка, продолговатый мозг, часть или весь IV желудочек и мост. Критерий на МРТ (Greenberg, 1997) – середина IV желудочка ниже линии, соединяющей бугорок турецкого седла и внутренний затылочный гребень. Характерен kink-симптом (перекручивание, петля).
Сочетается с другими дизрафиями. Практически все больные имеют миеломенингоцеле. Может быть spina bifida occulta, стеноз водопровода, гидроцефалия, маленькая ЗЧЯ, недоразвитие серпа, дисгенензия мозолистого тела. Операции: шунтирование, в случае успеха — декомпрессия ЗЧЯ. Практически все дети с пороками развития Киари II также страдают от расщепления позвоночника с сопутствующим миеломенингоцеле.
Киари III типа
Грыжевое выпячивание всего содержимого ЗЧЯ через spina bifida на уровне С1/С2 или в спинной канал. Крайне редкое состояние. Обычно несовместимо с жизнью.
Сопутствующие изменения
Аномалия Киари I сочетается:
- гидромиелией
- синдромом Клиппеля-Фейля
- гидроцефалией
- асиммиляцией атланта
- базиллярной импрессией

Рис.4

Рис.5
Дифференциальная диагностика
- дистопия мозжечковых миндалин (тонзиллярная эктопия),
- грыжа миндалин мозжечка (отек мозга и вклинение миндалин мозжечка в большое затылочное отверстие),
- спонтанная внутричерепная гипотензия.
Лечение
Оперативное лечение — декомпресионная остеотомия большого затылочного отверстия. При наличии гидроцефалии — дренаж желудочковой системы.

Рис.3
Полная или частичная перепечатка данной статьи, разрешается при установке активной гиперссылки на первоисточник
Автор: врач-рентгенолог, к.м.н. Власов Евгений Александрович
Похожие статьи
Источник
Аномалия Арнольда-Киари — это отклонение в развитии. И конкретно отклонение касается связки головного и спинного мозга. Головной мозг переходит в спинной на уровне большого затылочного отверстия. Четкой границы перехода нет.
Однако расположение ствола мозга относительно костей черепа и шейного отдела позвоночника по разным причинам может меняться. Это несовпадение приводит к сжатию спинного мозга в районе шейного отдела. Нормальная циркуляция спинномозговой жидкости нарушается.
Эта ситуация еще называется мальформация. Название происходит от латинских malus (что означает плохой) и formation (образование). Проще говоря возникает аномалия развития, за которой стоят изменение строения и функций.
В данном случае имеет место мальформация Арнольда-Киари в месте перехода черепа в позвоночник, то есть мальформация головного мозга.
Описали эту патологию австрийский патологоанатом Ханс Киари (в 1891 году) и немецкий патологоанатом Юлиус Арнольд (в 1894 году). Отсюда и сложное название.
Статистика указывает на то, что частота заболевания не такая уж и редкая — до 8.4 заболевших на 100 тысяч населения. Дополнительно к аномалии (до 80% случаев) выявляется сирингомиелия (образование кист в ткани спинного мозга).
Аномалия Арнольда-Киари — что это
Справочно. Аномалия Арнольда-Киари — это нарушение развития, которое приводит к опущению мозжечковых миндалин ниже анатомического уровня и к ущемлению продолговатого мозга.
Нормой считается расположение миндалин мозжечка выше большого затылочного отверстия. В процессе мальформации они могут сместиться даже до уровня второго шейного позвонка. При таком смещении усиливается блокировка тока спинномозговой жидкости.
Не всегда удается распознать это заболевание сразу, а затем происходит его резкая манифестация. Проявление патологии происходит к 25 — 40 годам.
При обнаружении характерных для аномалии симптомов необходимо обратиться к врачу, в противном случае риск развития инфаркта спинного мозга значительно возрастает.
Поскольку речь идет об отклонении от нормального развития организма, заболевание часто называют мальформация Арнольда-Киари.
Особенности мальформации
Справочно. Задний отдел мозга перемещается к большому затылочному отверстию (БЗО), когда у черепной ямки оказываются слишком малые размеры. В итоге формируется порок, возникают нарушения циркуляции ликвора.
Кости черепа не позволяют БЗО менять свой диаметр, поэтому при любых смещениях мозговых структур ущемляются близлежащие ткани. Последствия такого явления могут носить для пациента фатальный характер.
Продолговатый мозг отвечает за работу сердечно-сосудистой и дыхательной системы организма. Его сдавливание приводит не только к неврологическому дефициту, но может давать более опасные последствия, вплоть до смерти больного.
Смещение правого и левого полушарий мозжечка останавливает движение ликвора, что провоцирует гидроцефалию. Водянка повышает риск осложнений тех расстройств, которые уже имеются у пациента.
Процент пациентов с врожденной формой аномалии Киари невелик.
Справочно. Последние данные говорят о приобретенном характере. Дистопия миндалин мозжечка обусловлена быстрым ростом тканей при медленно протекающих изменениях в строении черепа. В медицине также известна под названием эктопия миндалин мозжечка.
Синдром Арнольда-Киари без проявления симптоматики может случайно обнаружиться во время проведения МРТ.
Причины развития аномалии
Мнения медиков о причинах появления аномалии расходятся. Есть несколько теорий, объясняющих то, каким образом развивается порок.
Неврологи выделяют две патологии, приводящие к формированию мальформации Киари (Chiari malformation):
- Развитие плода в утробе матери может пойти с нарушением — черепная ямка окажется меньше анатомической нормы, отделы мозга приобретут обычные параметры.
- Размеры отделов увеличены, при этом параметры задней черепной ямки и БЗО отвечают нормам. Увеличивающийся мозг устремляется к отверстию.
Врожденная аномалия у ребенка развивается, если беременная женщина не контролирует прием лекарств, употребляет алкоголь, курит на ранних сроках беременности.
Кроме того, вирусные инфекции у будущей матери (краснуха, цитомегаловирус) могут пагубно сказаться на развитии плода.
Справочно. Возникновению заболевания могут предшествовать различные родовые травмы, гидроцефалия, сильные повреждения головы у взрослого человека.
Типы аномалии
Рассматриваются четыре типа. Классификация производится с ориентацией на определенные основания.
Существенными признаками оказываются следующие изменения: те, что произошли в головном мозге на структурном уровне, те, что говорят о недоразвитости черепной коробки.
Аномалия Арнольда-Киари 1 типа отличается сдвигом мозжечковых миндалин, сопровождается нарушением циркуляции ликвора.
Последний заполняет узкий канал спинного мозга, вызывая гидромиелию. Указанный тип аномалии носит благоприятный прогноз. Он часто диагностируется у подростковой и взрослой групп населения.
Аномалия Арнольда-Киари 2 типа проявляет себя у новорожденных детей. Здесь наблюдается еще большее смещение отделов. Помимо мальформации, у грудничков диагностируется спинномозговая грыжа, обнаруживается аномальное развитие позвоночного столба.
В области затылка происходит выпячивание мозгового вещества через мягкую оболочку, мозжечок оказывается там же. Такова картина аномалии Арнольда-Киари 3 типа.
Внимание. Аномалия Арнольда-Киари 4 типа дает о себе знать тем, что мозжечок новорожденного оказывается недоразвитым, не занимает должного анатомического положения. Такая патология делает младенца неприспособленным к жизни, летальный исход неизбежен.
Степени тяжести
Сколько живут с аномалией Арнольда-Киари 1-й степени? Такой вопрос часто задают люди, услышавшие свой диагноз. Такая степень тяжести является самой невысокой, клинические проявления могут не отмечаться.
Спровоцировать возникновение симптомов могут черепно-мозговые травмы и повреждения позвоночника в верхней его части. Также запустить процесс может развившаяся нейроинфекция.
Аномалии 2 и 3-й степени уже сопровождаются патологическими изменениями в нервной ткани. У больного часто обнаруживают:
- смещение мозгового вещества;
- кисты проводящих ликвор путей;
- недоразвитость некоторых извилин мозга;
- гипоплазию подкорковых узлов.
Аномалия Арнольда-Киари — симптомы
Говоря о симптомах аномалии, надо, прежде всего, различать вариационные ее типы. Первый тип мальформации сопровождается несколькими синдромами, среди которых: гипертензионный, церебеллярный, сирингомиелический, бульбарный и т.д.
Гипертензионный синдром представляет собой повышение давления внутри черепа (ВЧД). Характерными симптомами будут интенсивные затылочные боли, тошнота, рвота, ригидность шейных мышц.
Церебеллярный синдром характеризуется речевыми расстройствами, нарушениями двигательной функции. При этом отсутствует четкость движений, затруднена мелкая моторика.
Сирингомиелический синдром проявляется потерей чувствительности в конечностях. Больной может получить случайный ожог, не заметив этого сразу. При обследовании обнаруживаются кисты спинного мозга.
Другие типы аномалии сопровождаются более тяжелой симптоматикой.
Справочно. У новорожденных страдает дыхание, возможна его остановка, отмечаются нарушения в глотании. Ребенок не может полноценно питаться. Посинение кожных покровов, гипертонус мышц, нистагм — вот основные проявления аномалии этих типов.
Возможные осложнения
Мальформация в некоторых случаях провоцирует достаточно опасные осложнения и может привести многих пациентов к инвалидности. Часто отмечаются увеличение ВЧД, дыхательные расстройства, апноэ, на фоне мальформации развиваются инфекционные заболевания легких и мочеполовой системы.
Справочно. Тяжело протекающая патология может стать причиной наступления комы, остановки в работе сердца, в итоге, быстрой смерти.
В запущенных случаях реанимация позволяет лишь поддерживать жизненно важные функции, сдавленный мозг восстановить практически невозможно.
Постановка диагноза
Осмотр невролога, сбор анамнеза представляют собой часть диагностики — они необходимы, но недостаточны.
Энцефалограмма, диагностика нарушения кровообращения в головном мозге и шейном отделе позвоночника могут косвенно показать наличие ВЧД.
С помощью рентгенографии, компьютерной томографии можно зафиксировать дефекты формирования черепной коробки. Но для определения состояния нервной ткани такие способы окажутся малоинформативными.
Справочно. МРТ сегодня считается единственно достоверным методом, позволяющим провести точную диагностику и своевременно распознать синдром Арнольда-Киари.
Процедура предполагает полное обездвиживание пациента, чего легко добиться от взрослого человека. Трудности возникают, когда пациентами являются маленькие дети. В этом случае необходимо применение общего наркоза.
Варианты лечения
После постановки диагноза лечение больного осуществляет нейрохирург или невролог. В исключительных случаях устранение аномалии возможно лишь путем проведения операции.
Если единственный симптом болезни — головная боль, то врачи ограничиваются медикаментозной терапией. Специалисты подбирают препараты:
- устраняющие воспаление (Найз, Ибупрофен, Диклофенак);
- анальгетики (Кеторол);
- спазмолитики (Мидокалм).
Справочно. Показанием к операции является сильное сдавливание отделов мозга, явно выраженные неврологические расстройства, если положительный эффект от приема лекарственных препаратов не наблюдается.
Оперативное вмешательство
Благодаря хирургическому вмешательству можно устранить чрезмерное давление на нервную ткань и привести в норму движение ликвора. Одной из проводимых операций является краниовертебральная декомпрессия, направленная на увеличение размеров задней черепной ямки.
Внимание. Декомпрессия относится к классу травматичных и рискованных операций. Согласно статистике, она приводит к осложнениям у каждого десятого пациента.
Риск летального исхода у прооперированных больных выше, чем у тех, кто операцию не переносил. Нейрохирурги стараются проводить такой тип вмешательства только в самых крайних случаях, когда налицо явные признаки сдавливания мозга.
Иной вариант устранения последствий мальформации предполагает шунтирование, которое способно обеспечить отвод ликвора из черепной коробки. Благодаря имплантации специальных трубок жидкость перетекает в грудную и брюшную полости, ВЧД снижается.
В наиболее острых случаях требуется немедленная госпитализация больного и проведение всего спектра терапевтических, профилактических и коррекционных процедур.
Прогноз выживаемости
Продолжительность жизни зависит от типа аномалии и степени ее тяжести. Первый тип позволяет сделать благоприятный прогноз, поскольку симптоматика либо вовсе отсутствует, либо возникает после получения травм головы.
Если никаких проявлений болезни нет, то продолжительность жизни больных такая же, как и у здоровых людей.
Для пациентов со вторым типом аномалии прогноз хуже, она переносится тяжелее.
Иногда борьба с очаговой неврологической симптоматикой не приносит плодов даже при активном лечении медикаментами. В таком случае требуется хирургическое вмешательство, чтобы впоследствии изменения по неврологической части были менее выраженными.
Справочно. Третий и четвертый типы мальформации являются самыми тяжелыми для пациентов, прогноз зачастую неблагоприятен.
Заболевание затрагивает важные структуры мозга, у больного отмечаются пороки внутренних органов. Часто функции ствола мозга страдают настолько, что нарушения оказываются несовместимыми с жизнью.
Источник