Болезнь крейтцфельдта якоба на мрт

Диагностика болезни Крейтцфельдта-Якоба по КТ, МРТ, ПЭТ головного мозгаа) Терминология: б) Визуализация: 1. Общие характеристики болезни Крейтцфельдта-Якоба (БКЯ):
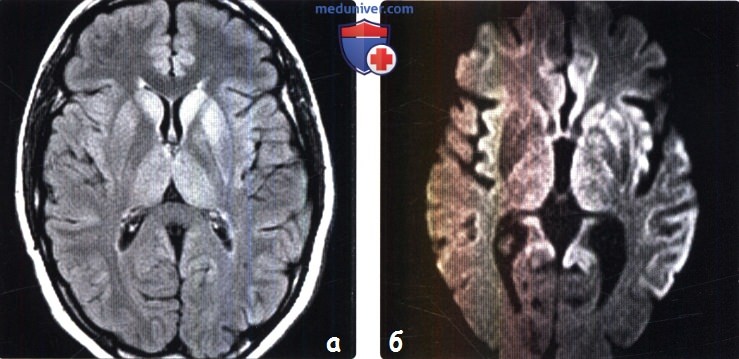
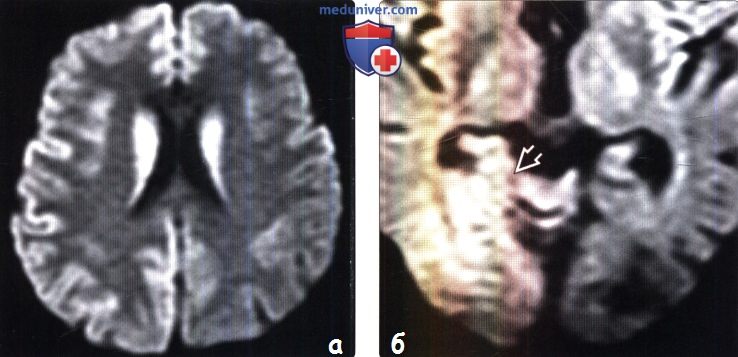
2. КТ признаки болезни Крейтцфельдта-Якоба (БКЯ): 3. МРТ признаки болезни Крейтцфельдта-Якоба (БКЯ): 4. Радионуклидная диагностика болезни Крейтцфельдта-Якоба (БКЯ): 5. Рекомендации по визуализации:
в) Дифференциальная диагностика болезни Крейтцфельдта-Якоба (БКЯ): 1. Гипоксически-ишемическое поражение: 2. Синдром осмотической демиелинизации: 3. Синдром Лея: 4. Другие причины деменции: 5. Кортикобазальная дегенерация: 6. Болезнь Вильсона: 7. Атеросклероз:
г) Патология: 1. Общие характеристики болезни Крейтцфельдта-Якоба (БКЯ): 2. Макроскопические и хирургические особенности: 3. Микроскопия:
д) Клиническая картина: 1. Проявления болезни Крейтцфельдта-Якоба (БКЯ): 2. Демография: 3. Течение и прогноз: 4. Лечение: е) Диагностическая памятка: ж) Список литературы: — Также рекомендуем «Болезнь Паркинсона на МРТ головного мозга» Редактор: Искандер Милевски. Дата публикации: 4.5.2019 |

Источник
Ср, 04/05/2011 — 23:47
#1

Не на сайте
Был на сайте: 1 год 7 месяцев назад
Зарегистрирован: 22.03.2008 — 22:15
Публикации: 54888
О. Хабиб
O. Habib
Губчатые энцефалопатии представляют собой инфекционные дегенеративные заболевания. Некоторые семейные формы обусловлены генетически. В ткани мозга обнаруживают патологические белковые частицы — прионы (PrPres), которые представляют собой устойчивую патологическую (лишенную нуклеиновой кислоты) копию прион-протеина (PrP), присутствующего в здоровой нервной ткани всех млекопитающих. PrP — это мембранный сиалогликопротеин, состоящий из 253 аминокислот; он имеет двухмерную пространственную структуру и кодируется единственным геном на коротком плече хромосомы 20. Патологическая форма белка обнаруживается только в пораженной инфекционной губчатой энцефалопатией (ИГЭ) ткани мозга. PrPres и PrP имеют различные пространственные структуры и свойства: PrPres нерастворим в воде, нечувствителен к протеазам, имеет в структуре большее количество b-цепей и меньшее количество a-спиралей. Предполагают, что конверсия PrP в PrPres происходит во время транскрипции. Пока неясно, является ли PrPres одновременно патогенным и вирулентным (так как не обнаружена нуклеиновая кислота, а в эксперименте удалось получить PrPres из PrP в ацеллюлярной системе) или в PrPres включен некий вирус (т.е., имеется нуклеиновая кислота), который и обусловливает инфекционную передачу, а PrPres дает только токсический эффект. Существование частицы с независимым геномом наиболее удачно объясняет наличие нескольких разновидностей PrPres. Одна из гипотез предполагает существование пока не распознанной молекулы, которая, прикрывая белок и вирус, мешает «увидеть» нуклеиновую кислоту.
Генетическая предрасположенность. Гену, ответственному за синтез PrP, свойствен полиморфизм. 129-ый Кодон может кодировать либо метионин, либо валин. 50% населения гетерозиготны по кодону 129 (метионин/валин), 40% гомозиготны по метионину и 10% — по валину. А поскольку все больные ИГЭ — гомозиготы, то утрату полиморфизма можно считать фактором, предрасполагающим к возникновению заболевания.
Разновидности PrPres. У пораженных ИГЭ овец верифицируют до 20 разновидностей PrPres и соответственно наблюдают различные инкубационные периоды и характер поражения ЦНС. Для ИГЭ коров характерен только один вид PrPres. Для болезни Крейцфельдта — Якоба (БКЯ) известны 4 разновидности (по иммуноблоттингу) PrPres.
Разновидности БКЯ. БКЯ является одним из видов человеческих ИГЭ. Помимо БКЯ к этой группе заболеваний относятся болезнь Куру («смешливая» болезнь, эндемична для Новой Гвинеи, характерна 100% летальность), болезнь Гертсмана — Штройслера — Шайнкера (Gertsmann Straussler Scheinker) и фатальная семейная бессонница. По клинической картине и способу передачи различают следующие разновидности: классическая/спорадическая, семейная/генетическая, ятрогенная и британская ИГЭ.
Классическая форма описана Крейтцфельдтом (1920 г.) и Якобом (1921 г.). Заболеваемость в мире составляет 0,5-1 случай на 1 млн человек в год; во Франции регистрируется 30-50 случаев в год; 90% всех случаев БКЯ приходится на классическую форму. Заболевание обычно дебютирует в 55-75 лет, за 70 лет со времени описания зарегистрировано 20 исключений- ювенильных случаев (16-40 лет), которые рассматриваются сейчас как британский вариант. Характерно постепенное прогрессирование симптомов поражения ЦНС, в 13% случаев заболевание «разворачивается» в течение нескольких дней.
Клиническая картина: начальные симптомы в 35% случаев — депрессия и расстройство ментальных функций, в 34% — неврологические симтомы с преимущественным поражением зрительных функций и мозжечка; в 21% — сочетанные. Симптомы быстро прогрессируют: развивается глубокая деменция, часто в сочетании с мутизмом. Атаксия, тремор и ригидность обусловливают локомоторные расстройства вплоть до иммобилизации. Наступает смешанная кортико-ретинальная слепота; на поздней стадии нередки пирамидные расстройства, миоклонии и эпилептические припадки. Смерть наступает до истечения года с момента появления первых симптомов, в 90% случаев — к 5-му месяцу. Параклинические данные не специфичны: на ЭЭГ- периодические судорожные разряды, ликвор в норме (PrPres пока не определяют), на компьютерной томограмме — атрофия коры полушарий и мозжечка. Патоморфологически определяют уменьшение количества кортикальных нейронов, вакуолизацию ткани мозга, который приобретает вид губки; глиальную, в основном астроцитарную, пролиферацию. Признаки воспаления отсутствуют. PrPres верифицируют с помощью иммуногистохимического анализа и иммуноблоттинга. С 1968 г. инокулируют экстракт ткани мозга восприимчивым животным (приматам, грызунам, кошкам). Диагноз ставится на основании клинических проявлений и подтверждается патоморфологическими данными и заражением лабораторных животных.
Эпидемиология. Несмотря на обилие данных, пока не удается объяснить происхождение классической формы. Некоторое время назад обсуждалось алиментарное заражение в связи с увеличением заболеваемости в Исландии. Дискуссия возобновилась после верификации ИГЭ коров в 1986 г. Для заболевших людей характерны повышенный травматизм и частые хирургические вмешательства в анамнезе. Не исключено, что представители некоторых профессий — пастухи, фермеры, медики — подвержены повышенному риску заболевания.
Семейная (генетическая) форма составляет 5-6 % всех случаев БКЯ, наследуется по аутосомно-доминантному механизму. Чаще всего это мутация — замена аспартатовой кислоты на аспарагин в 178-й позиции молекулы PrP. Заболевание дебютирует на 5-10 лет раньше, чем при классической форме, течение заболевания такое же. Существуют изолированные этнические группы, где заболеваемость БКЯ в 30-100 раз выше, чем для населения в целом, что обусловлено особенностями генотипа и специфичностью мутаций. Это — ливийские евреи (Средний Восток и Средиземноморье), словацкие и чилийские общины, в которых распространены внутрисемейные браки.
Ятрогенная БКЯ появилась в последние годы, случаев мало, но очевиден их драматизм в психологическом, этическом и медико-юридическом аспектах. По типу заражения различают две группы. Центральное заражение происходит во время операций, хирургических манипуляций, через трансплантаты и хирургический инструмент. Известны 24 случая заражения при пересадке взятой у погибших от БКЯ твердой мозговой оболочки, 1 случай — при пересадке роговицы, 2 случая — после стереотаксической операции. Инкубационный период после инокуляции — от 7 до 120 мес. Закономерен вывод, что «инфекционное начало» (вирус?) очень устойчиво к воздействию традиционных антисептиков, которым обрабатывают инструменты и трансплантаты. Следовательно, они должны быть надежно обеззаражены, а оптимальным выходом является использование одноразовых инструментов. Известные случаи периферического заражения связаны с введением СТГ. Из всех 50 зарегистрированных 70-80% приходятся на Францию. 4 случая в Австралии связаны с введением ГТГ гипофиза для индукции овуляции. Сегодня очевидно, что замена естественного СТГ на рекомбинантный ( продукт генной инженерии) полностью устранит риск. Латентный период составляет 18-28 лет. Клиническая картина больше похожа на симптоматику болезни Куру — неврологические симптомы преобладают над интеллектуальной деградацией. У большинства больных наблюдают утрату полиморфизма на 129-м кодоне и характерное «поведение» прийона при выполнении иммуноблоттинга. В эксперименте доказана возможность заражения животных кровью больных. Таким образом безопасность донорских тканей и органов должна быть обеспечена, а их использование — обосновано.
Британский вариант. С 1995 по 1997 г. в Великобритании зарегистрировано 14 случаев БКЯ у молодых людей (и еще один во Франции). Общими для всех случаев были: нормальный ген PrP, гомозиготность по кодону 129, отсутствие каких-либо факторов риска, пространственно-временное совпадение (инкубация в несколько лет), характерное «поведение» PrPres при проведении иммуноблоттинга, напоминающее таковое при ИГЭ кошек, зараженных алиментарно. Появилась гипотеза о возможности алиментарного заражения при употреблении мяса больных животных. Ситуация прояснится в ближайшие два года с завершением экспериментов на трансгенетических мышах. Не исключено обнаружение неизвестных ныне защитных факторов.
Литература:
Beauvais P. La maladie de Creutzfeldt-Jakob. La plus important des maladies a prion. La Presse Medical 1997;26:3787-92.
Ср, 04/05/2011 — 23:53
#2
Не на сайте
Был на сайте: 1 год 7 месяцев назад
Зарегистрирован: 22.03.2008 — 22:15
Публикации: 54888
Крейтцфельдта — Якоба болезнь (Н.G. Creutzfeldt, немецкий невропатолог и психиатр, 1885-1964; A. Jakob, немецкий невропатолог, 1884-1931; синоним: кортикостриоспинальная дегенерация, спонгиоформная энцефалопатия, спастический псевдосклероз) — прогрессирующее заболевание центральной нервной системы с преимущественным поражением коры головного мозга, подкорковых узлов и спинного мозга. Большинство исследователей включает болезнь Крейтцфельдта — Якоба болезнь в группу медленных вирусных инфекций. Определенную роль в развитии заболевания играет генетическая предрасположенность, о чем свидетельствуют высокая частота семейных случаев заболевания и поражение близнецовых пар.
Патогенез не известен. Установлено, что при этом заболевании в коре головного мозга снижена активность ряда ферментов, особенно лактатдегидрогеказы и ДРН-липоамид-дегидрогеназы, что указывает на нарушение окислительных процессов. Отмечается также общее снижение содержания липидов в ткани головного мозга, особенно в сером веществе.
При морфологическом исследовании головного мозга выявляются уменьшение его массы и объема, сглаженность извилин, выраженное расширение боковых желудочков, побледнение мягкой мозговой оболочки. Микроскопическая картина характеризуется дегенеративными изменениями нервных клеток и пролиферацией нейроглии в обширных участках коры, подкорковых образованиях, мозжечке, стволе мозга и спинном мозге. Демиелинизация проводящих путей и ассоциативных волокон носит вторичный характер по отношению к дегенеративным изменениям в нервных клетках. Чаще она наблюдается в области внутренней капсулы, передней спайки, мозолистого тела.
Клинические признаки Крейтцфельдта — Якоба болезни обычно появляются в возрасте между 40-70 годами, однако описаны случаи начала заболевания в молодом возрасте. Различают нисходящий и восходящий типы развития заболевания. При нисходящем типе ранние симптомы представлены расстройствами корковых функций — снижением памяти, внимания, нарастающей деменцией, апраксией, моторной афазией, слуховыми и зрительными галлюцинациями, зрительной агнозией, нарушениями поведения (апатией, беспокойством, раздражительностью, депрессией, эйфорией, агрессивностью и др.), Менее постоянны сенсорная афазия, аграфия, алексия, нарушения схемы тела. При относительно редком восходящем типе вначале появляются спастические и (или) вялые параличи конечностей (монопарезы, парапарезы, тетрапарезы), тазовые расстройства (недержание мочи, кала или их задержка). Затем присоединяются расстройства координации движений, туловищная атаксия, гиперкинезы в виде миоклоний, хореоатетоза, реже атетоза и тиков, бульбарная, псевдобульбарная мозжечковая или экстрапирамидная дизартрия. Патология черепных нервов проявляется глазодвигательными расстройствами, дефектом полей зрения. слабостью иннервации мышц лица, бульбарными и псевдобульбарными нарушениями. Снижение болевой, температурной, тактильной и глубокой чувствительности наблюдается чаще в ногах, реже — в руках, в области туловища и головы. Более редкими расстройствами чувствительности являются сильные боли и парестезии. Мышечный тонус меняется в зависимости от преимущественной локализации очагов поражения. При поражении периферического мотонейрона развивается гипотония мышц; повреждение центрального двигательного пути приводит к спастичности мышц, при поражении подкорковых узлов может отмечаться паркинсоноподобная мышечная ригидность. Сухожильные рефлексы повышены или снижены. Брюшные рефлексы обычно снижены. В поздней стадии заболевания вызываются рефлексы орального автоматизма — сосательный, хоботковый, Маринеску — Радовича и др.
Диагноз основан на данных дополнительных исследований. Характерны изменения на ЭЭГ в виде вспышек волн в лобно-височных областях с последующим сжижением вольтажа биоэлектрической активности. Часто отмечается угнетение альфа-ритма. При эхоэнцефалографии выявляют расширение желудочков мозга, на пневмоэнцефалограммах — расширение желудочков мозга и субарахноидальных пространств, на рентгеновских компьютерных томограммах мозга — расширение желудочков мозга, атрофию мозгового вещества, уменьшение объема мозга. Дифференциальный диагноз проводят с пресенильной деменцией, Альцгеймера болезнью, Пика болезнью, Гентингтона хореей, гепатоцеребральной дистрофией, миоклонус-эпилепсией, лейкоэнцефалитом.
Вс, 19/06/2011 — 17:29
#3
Не на сайте
Был на сайте: 1 год 7 месяцев назад
Зарегистрирован: 22.03.2008 — 22:15
Публикации: 54888
КРЕЙТЦФЕЛЬДТА — ЯКОБА БОЛЕЗНЬ.
Втр, 10/04/2012 — 17:07
#4
Не на сайте
Был на сайте: 1 год 7 месяцев назад
Зарегистрирован: 22.03.2008 — 22:15
Публикации: 54888
Крейтцфельдта — Якоба болезнь
Пнд, 06/05/2013 — 21:50
#5
Не на сайте
Был на сайте: 1 год 7 месяцев назад
Зарегистрирован: 22.03.2008 — 22:15
Публикации: 54888
Пт, 09/03/2018 — 19:38
#6
Не на сайте
Был на сайте: 1 год 7 месяцев назад
Зарегистрирован: 22.03.2008 — 22:15
Публикации: 54888
Источник