Цитотоксический отек головного мозга мрт

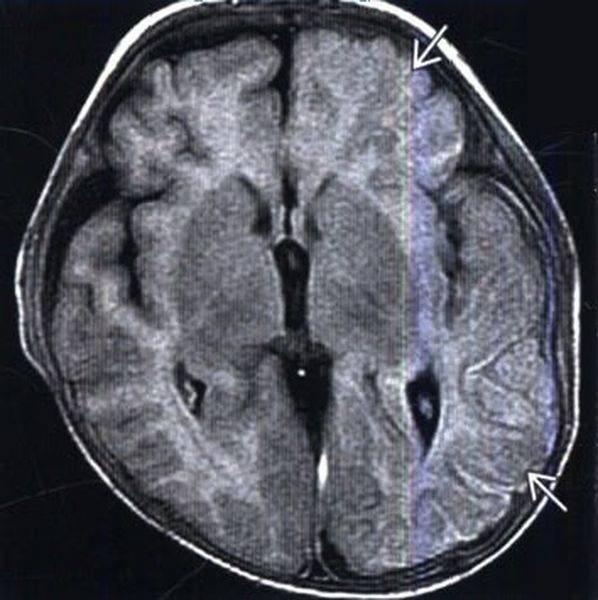 Посттравматический отек левого полушария (показано стрелками)
Посттравматический отек левого полушария (показано стрелками)Отек головного мозга – увеличение объема тканей, возникающее вследствие постепенного или стремительного накопления жидкости, изменения структур и функциональности клеток. Патологическое явление является осложнением ряда заболеваний. Опасность отека и набухания состоит в том, что церебральные структуры оказываются сдавлены черепом и отрогами твердой оболочки и не могут дальше увеличиваться в объеме. Данный процесс влечет за собой повышение внутричерепного давления, компрессию сосудов, дислокацию мозговых структур, нарушение кровообращения и, в конечном счете, — гибель нейронов. Смерть от отека мозга, если его не лечить, неизбежна. Вовремя проведенное обследование может помочь сохранить жизнь человеку.
Магнитно-резонансную и компьютерную томографии широко используют для быстрой диагностики патологических явлений в головном мозге. Обсуждаемый процесс может иметь несколько форм: цитотоксическую или вазогенную. Каждый вид отличается причинами возникновения и локализацией патологических проявлений.
Наиболее распространён вазогенный тип отека. Характеризуется переходом жидкости из сосудов в белое мозговое вещество. Патология возникает в связи с нарушением работы гематоэнцефалического барьера. Данный тип отека наблюдают вокруг опухолей (первичных и вторичных), при абсцессах, инсультах, ушибах, кровоизлияниях и пр. Подвидом рассматриваемого осложнения считают интерстициальный вариант, который возникает вследствие повышения давления в желудочках, что приводит к пропотеванию спинномозговой жидкости в интерстициальное пространство. Данный процесс вызывает отек белого вещества. Частыми причинами являются обструктивная гидроцефалия и менингит.
Цитотоксический (клеточный) вид патологии возникает в сером веществе, может быть вызван интоксикацией, отравлением, ишемическим инсультом, вирусными инфекциями, травмами головы и т.д. При данной форме отека на первом этапе не происходит повреждение гематоэнцефалического барьера, наблюдают изменение ионного баланса на поверхности клеточных мембран.
Любой из описанных вариантов патологии крайне непродолжительное время может существовать изолированно. Принято рассматривать отек и набухание головного мозга как звенья одного патологического процесса.
С помощью МРТ возможно определить преобладающий вариант, что обуславливает выбор схемы лечения.
Признаки отека головного мозга
 Снимки головного мозга на МРТ
Снимки головного мозга на МРТ
Основной симптом, по которому врачи безошибочно определяют отек и набухание головного мозга – расстройство сознания от легких до тяжелых форм.
На начальной стадии при медленном прогрессировании церебрального процесса больной остается в адекватен и ориентирован в себе, месте и времени, могут развиваться судороги. Выделяют следующие общие признаки, указывающие на возможный отек мозга:
- сильная головная боль, сопровождающаяся тошнотой и рвотой, особенно в утренние часы;
- нарушение двигательных функций, чувствительности, зрения, координации и т.п.;
- головокружение;
- галлюцинации;
- судороги;
- психомоторное возбуждение;
- нарушение вегетативных и витальных функций;
- панические атаки и пр.
Последние признаки из списка – самые опасные, так как сопровождают компрессию ствола мозга и требуют неотложной медицинской помощи.
При возникновении любых вышеуказанных настораживающих проявлений при неврологических заболеваниях или опухолях мозга и головы следует незамедлительно обращаться к врачу. После диагностики менингеальных симптомов и признаков нарушения сознания будет назначено соответствующее обследование на МРТ или КТ.
Отек мозга на МРТ, как выглядит?
Вазогенный отек чаще всего возникает вследствие опухоли, абсцесса. На МРТ регистрируют гиперинтенсивный сигнал в режиме Т2 взвешенного изображения и FLAIR (с подавлением сигнала свободной воды) без ограничения диффузии.
Вазогенный отек из-за абсцесса на МРТ
Цитотоксический отек головного мозга на МРТ невозможно определить на Т2 или Т1 режимах, так как процесс происходит из-за перераспределения воды из вне- во внутриклеточное пространство. Патологию определяют за счет соизмеримого снижения диффузии, которая проявляется повышенным сигналом на ДВИ (диффузионно-взвешенных изображениях). Данные изменения возможно выявить в подострой фазе (до 14 дней).
Пройти МРТ головы по назначению врача Вы можете в медицинском центре «Магнит». Для выбора оптимальной даты, времени процедуры заполните обратную форму связи на сайте, после чего наши консультанты свяжутся с Вами.
Источник
Отек головного мозга вызван различными патологическими состояниями. Цитотоксический отек обусловлен двумя отдельными патогенетическими процессами с различными молекулярными и физиологическими механизмами: связанными с цитотоксическим (клеточным) отеком нейронов и астроцитов и связанными с транскапиллярным потоком Na + и других ионов, воды и макромолекул сыворотки крови. По большому счету , цитотоксический отек возникает из-за неконтролируемого или точнее нескомпенсированного притока катионов, главным образом Na +, через катионные каналы.
Цитотоксический отек может быть результатом почти любого поражения мозга, включая травму ( травма головного мозга ежегодно фиксируется у 1,4 миллиона человек в США , что приводит к 50 000 смертей и 235 000 госпитализаций) , инфаркт, инсульт ( В США инсульт является третьей по распространенности причиной смерти, причем ежегодно отмечается более 730 000 случаев заболевания.) новообразование, абсцесс или такие состояния, как гипоксия или токсическое воздействие или метаболическое нарушение. Цитотоксический отек определяется как клеточный процесс, иначе известный как клеточный отек, при котором внеклеточные Na + и другие катионы входят в нейроны и астроциты и накапливаются внутриклеточно, частично из-за отказа энергетически зависимых механизмов экструзии. Неконтролируемый приток катионов происходит в основном через катионные каналы. Приток катионов, в свою очередь, приводит к притоку анионов, который поддерживает электрическую нейтральность, и в совокупности эти явления приводят к притоку воды, что приводит к осмотическому расширению клетки, то есть цитотоксическому отеку. Цитотоксический отек сам по себе не приводит к отеку мозга, но образование цитотоксического отека истощает внеклеточное пространство Na + , Cl -, и воды, тем самым создавая новый градиент для этих молекул через капилляр гематоэнцефалического барьера. При соответствующих изменениях проницаемости капилляров новый градиент, созданный цитотоксическим отеком, приводит к замедлению транскапиллярного образования ионного отека. Таким образом, цитотоксический отек имеет важное значение сам по себе, поскольку он сигнализирует о преморбидном клеточном процессе, который почти неизбежно приводит к гибели онкотических или некротических клеток, но тем не менее важно, цитотоксический отек обеспечивает своего рода «движущую силу» для образования ионного отека, представляющего собой процесс, который вводит новую массу (Na + , Cl — , H 2 O), в конечном итоге отвечающую за набухание мозга.
Когда поражение мозга приводит к ишемии или гипоксии, очень мало продуцируется новых АТФ вслдествие отмены окислительного фосфорилирования. Клетки быстро используют свои запасы АТФ, и, если нормоксия не будет восстановлена, нарушенное клеточное оборудование теряет способность поддерживать гомеостаз. Первичный активный транспорт, в основном АТФ-зависимая Na + / K + АТФаза требует непрерывных расходов АТФ, которая необходима для поддержания гомеостаза. По сути баланс между выживаемостью и смертью клетки определяется борьбой между активностью электро насоса и каналами, которые позволяют Na + проникать в клетки.
Выживаемость клеток требует, чтобы Na + непрерывно экструдировали из внутриклеточного пространства, поскольку оно имеет решающее значение для поддержания нормального объема клеток. Истощение АТФ сопровождается неконтролируемым притоком внеклеточных ионов, в основном Na + , внутрь по их электрохимическим градиентам. Приток ионов натрия в свою очередь приводит к притоку Cl — через хлоридные каналы, и, как следствие, увеличение внутриклеточной осмолярности приводит к притоку воды через каналы AQP .
Внеклеточная вода проникает внутрь клетки, что приводит к увеличению объема внутриклеточной жидкости за счет внеклеточного пространства. Морфологически этот процесс приводит к изменениям в поверхностной архитектуре мембраны с заметным образованием в ней пузырьков. На начальных стадиях цитотоксического отека гематоэнцефалический барьер является неповрежденным и в значительной степени непроницаемым для ионов и жидкостей, поэтому внеклеточные ионы и потери воды не пополняются. Таким образом, движение жидкости, связанное с образованием цитотоксического отека, не приводит к изменению общего объема мозга, несмотря на наблюдаемое увеличение размера клеток.
Клетки в сером и белом веществе подвержены воздействию цитотоксического отека. Клеточный отек начинается в течение 30 минут после окклюзии MCA, особенно вокруг капилляров и сохраняется до 24 часов после реперфузии и приводит к среднему уменьшению внеклеточного пространства от нормальных 20% до 4-10%. Астроцитарный отек гораздо более заметен, чем отек нейронов. Астроциты более склонны к патологическому отеку, чем нейроны, потому что они участвуют в выведении К + и глутамата, которые вызывают осмотическую перегрузку, что, в свою очередь, способствует притоку воды. Астроцитарная, но не нейронная NKCC активируется повышенным внеклеточным K +и приводит к набуханию клеток. Важное значение имеет здесь также экспрессия высокого уровня водного канала AQP4.
Когда компенсаторные механизмы, такие как ионные насосы в плазматической мембране, плохо выполняют свою работу или вообще не работают, отекшая клетка погибает. Этот путь к гибели клеток был назван онкозом ( от греческого слова «онкос», что означает набухание) von Recklinghausen, в частности, для описания клеточной смерти при отеке. Этот термин является более конкретным способом классификации клеточной гибели, чем менее точные термины «случайная гибель клеток» или «некроз». Гибель отченых клеток также отличается от их апоптотической гибели. На электронно-микроскопическом уровне различие между двумя путями, ведущими к гибели клеток, становится очевидным: онкоз приводит к характерной форме клеток, которые показывают заметное увеличение объема и представляет собой мембранное повреждение плазмоламмы и других мембрани органелл, а также потерю мембранных фосфолипидов и исчезновение пятнистых ядер на поздней стадии, напротив, апоптоз представляет собой инволюцию клеток и их сморщивание.
Экспериментальные данные показывают четко очерченную последовательность метаболических реакций ткани головного мозга на снижение кровотока. Область мозга, в которой кровоток отсутствует или измеряется менее 10 мл / 100 г (мозговая ткань) / мин, быстро и необратимо повреждается менее чем за 6 минут, образуя «ишемическое ядро». Эта инфарктная ткань окружена » полутенью» гипоксической, но живой ткани с кровотоком более 20 мл / 100 г (ткань головного мозга) / мин. Клетки в полутени подвергаются цитотоксическому отеку и другим изменениям, которые потенциально обратимы, если перфузия восстанавливается в течение первых нескольких часов после травмы мозга. Однако, если гипоксические условия сохраняются, в конце концов умирают клетки с цитотоксическим отеком, расширяя масштаб клеточной смерти глубже в паренхиму, чем первоначально вовлеченное ядро. Таким образом, полутень является основной терапевтической мишенью для профилактики ишемического инсульта и травмы.
Ряд исследований показал, что фармакологическое ингибирование ионных каналов, включая неселективные катионные каналы, уменьшает очаговое ишемическое повреждение в моделях ишемического инсульта. Неселективные катионные каналы отличаются от селективных катионных каналов их проницаемостью; ионные селективные каналы обычно проницаемы для одного катиона, такого как Na + , K + или Ca 2+ , тогда как неселективный катионный канал может пропускать поток любого одновалентного катиона или даже смесь одновалентных и двухвалентных катионов. Вероятно, эти каналы играют важную роль во вторичной травме в зоне «полутени».
Кислотно-чувствительные ионные каналы ( ASIC) являются членами недавно обнаруженного семейства ионных каналов эпителиального натриевого канала / дегенерина. Кислотно-чувствительные гены ионного канала кодируют протонированные катионные каналы как в центральной, так и в периферической нервной системе. К настоящему времени клонированы шесть различных субъединиц ASIC, которые кодируются четырьмя генами ASIC1-ASIC. Кислотно-чувствительные ионные каналы представляют собой катионные каналы с водородным ионами, которые активируются при понижении рН, но обычно неактивны при физиологическом рН . Все ASIC проницаемы для Na + и, в меньшей степени, для Ca 2+ и блокируются амилоридом. Активация этих каналов приводит к увеличению возбудимости клеток.
Каналы ASIC ASIC1a и ASIC2a привлекли научное внимание в контексте нейропротекции. Ишемия и гипоксия приводят к заметному снижению рН ткани из-за неконтролируемой генерации молочной кислоты, а ацидоз является важной детерминантой неврологического повреждения. Субъединица ASIC1a может быть ответственна за опосредованную ацидозом, нейрональную травму, не зависящую от глутамата. Вероятность открытия ASIC1a возрастает с уменьшением рН ниже 7,0, а активация составляет половину максимума при рН 6,2, находящейся в диапазоне рН, который, как полагают, имеет место в «полутени» и сердцевине инфаркта, особенно в контекст гипергликемии. Активация ASIC1a способствует растяжению мембраны, высвобождению арахидоновой кислоты, получению лактата, состояний, которые наблюдаются в пределах инфаркта в виде набухающих клеток, Ca 2+ -зависимый фосфолипазы активируются, и происходит приток Ca 2+
Активация ASIC1a in vitro приводит к увеличению внутриклеточного Ca 2+ и индуцирует зависяще от времени повреждение нейронов, которое возникает в присутствии блокаторов каналов Ca 2+ с напряжением и глутаматных рецепторов. В моделях ишемического инсульта грызунов in vivo интра церебро — вентрикулярное введение блокаторов ASIC1a амилорида и тарантула (псальмотоксин 1) до начала ишемии, а также «выбивание гена» ASIC1a предотвращает ишемическое поражение. Канал ASIC2a вызвал особый интерес, поскольку временная глобальная ишемия вызывает его экспрессию в мозге крысы, в том числе в нейронах гиппокампа и коры.
Канал NC Ca-ATP представляет собой обнаруженный канал катионов, который проводит все неорганические одновалентные катионы, но являеся непроницаемым для Ca 2+ и Mg 2+
Открытие этого канала требует наномолярного Ca 2+ на стороне цитоплазмы. Физиологические уровни АТФ внутриклеточно блокируют открытие канала NC Ca-ATP , тогда как истощение ATP вызывает открытие канала. Предполагается, что канал NC Ca-ATP состоит из порообразующих и регуляторных субъединиц. Регуляторная субъединица SUR1, такая же, как и для K- АТФ- каналов в клетках поджелудочной железы. Поскольку SUR1 участвует в регуляции канала, фармакологические агенты, которые влияют на канал S AT1 с регулируемым KP, также влияют на канал NC Ca-ATP . Таким образом, NC Ca-ATP канальное отверстие блокируется соединениями сульфонилмочевины, такими как толбутамид и глибенкламид, и активность каналов увеличивается диазоксидом.
Канал NC Ca-ATP не конститутивно экспрессируется, а выражается в ЦНС после гипоксии или травмы. Канал был впервые обнаружен в недавно выделенных реактивных астроцитах, полученных из гипоксической внутренней зоны глиотической капсулы. С тех пор он также был идентифицирован в нейронах из ядра ишемического инсульта. В крысиных моделях ишемического инсульта регуляторная субъединица SUR1 транскрипционно регулируется в нейронах, астроцитах и капиллярных эндотелиальных клетках.
Последствия открытия канала изучались в изолированных клетках, которые экспрессируют канал, путем истощения АТФ с использованием Na + азида или Na + цианида в дополнение к 2-дезоксиглюкозе или с использованием диазоксида для открытия канала без истощения АТФ. Эти обработки индуцируют сильный внутренний ток, который деполяризует клетку полностью до 0 мВ и индуцирует цитотоксический отек и клеточное кровоизлияние. Эти эффекты воспроизводятся без истощения АТФ диазоксидом. После этих обработок, клетки умирают преимущественно посредством онкоза, а не путем апоптоза. Влияние блокировки каналов с использованием глибенкламида изучалось in vitro в реактивных астроцитах, которые экспрессирует канал. В клетках, подвергнутых воздействию Na + азида, преднамеренно истощает АТФ, глибенкламид блокирует деполяризацию мембран, значительно уменьшает кровоизлияние, связанное с цитотоксическим отеком, и значительно снижает смертностьуровень онкотических клеток. В модели массивного ишемического инсульта со злокачественным отеком головного мозга, связанного с высокой смертностью (68%), глибенкламид уменьшал смертность и отек мозга (избыток воды) на 50%. В модели инсульта, вызванной тромбоэмболией с задержкой спонтанной реперфузии, глибенкламид уменьшал объем поражения в два раза, а его использование ассоциировалось с кортикальной активностью , связанной с улучшенным лептоменингом побочным кровотоком из-за снижения массового эффекта от отеков.
Семейство канала TRP получило свое название от его роли в фототрансдукции дрозофилы . Это семейство содержит более 50 членов, 28 из которых, как известно, выражены у млекопитающих. Эти каналы различаются по своим режимам активации. Некоторые каналы TRP являются конститутивно открытыми, а другие реагируют на различные стимулы, такие как pH, окислительно-восстановительное состояние, осмолярность, растяжение, напряжение и внутриклеточный Ca 2+ Некоторые из этих каналов являются селективными для Ca 2+ , а другие являются неселективными и проницаемыми для одновалентных и / или двухвалентных катионов. Белки TRP имеют тенденцию образовывать гетеромультимеры и могут проявлять взаимозависимую экспрессию.
Анализ промоутерных областей членов подсемейства TRPC и TRPM TRPC1-7 и TRPM1-8 показывает, что эти члены обладают несколькими консенсус-сайтами связывания для одного или нескольких фак. торов транскрипции, связанных с ишемическим инсультом, что указывает на их возможное участие в гипоксическом повреждении ЦНС.
Электронейтральный котрансперсер NKCC кодируется геном из семейства катион-хлоридных котранспортеров. Этот канал опосредует связанное движение Na + и / или K + с Cl — , с стехиометрией 1Na + : 2K + : 2Cl — . Активность этого транспортера связана с регуляторными ионными реакциями эпителиальных клеток глии, нейронов, эндотелия и сосудистого сплетения. Хотя обнаружены две изоформы, только NKCC1, «домашняя» изоформа канала NKCC, играет роль в секреции и абсорбции натрия, регулировании объема клеток и поддержании внутриклеточной Cl -концентрации в ЦНС. Петлевые диуретики, такие как буметанид, могут ингибировать канал. Изоформа NKCC1 участвует во вторичном переносе неорганических ионов. Движущей силой потока ионов происходит в Na + градиент , создаваемого Na + / K + -АТФазы, с важным вкладом Cl -градиента в эпителиальных клетках. Котранспортер NKCC требует, чтобы все три иона (Na + , K + и Cl — ) одновременно присутствовали на одной стороне мембраны. Уменьшение внутриклеточного Cl — , гипертонический стресс, повышение внутриклеточного Са 2+ , и β-адренергич еские рецепторы стимуляция приводят к фосфорилированию NKCC1, что увеличивает активность канала. Киназы и фосфатазы способствуют регулированию NKCC1 через их противоположные эффекты.
Изоформа NKCC1 играет важную роль в поддержании физиологических внутриклеточных уровней концентрации Na + . Однако в патологических ситуациях, таких как ишемия и гипоксия, было показано, что это способствует чрезмерному притоку Na + , что приводит к цитотоксическому отеку. Данные in vitro в нейронах показывают, что потеря Cl — является достаточным и необходимым стимулом активации NKCC1. Аналогичным образом , работа этой изоформы каналов в астроцитах показывает повышенные уровни внеклеточного К + , чтобы они стали достаточными и необходимыми для активации астроцитов NKCC1. Генетическая абляция NKCC1, а также ее блок с помощью буметанида приводят к уменьшению внутриклеточного Cl — в гипоксических нейронах и блокирует клеточные проявления Na + — и Cl — в астроцитах. Исследования in vivo показали, что внутримозговый буметанид, вводимый через микродиализный зонд, до или во время ишемии / гипоксии, вызванной временной окклюзией MCA, является нейропротективным, улучшает повреждение головного мозга и уменьшает отек мозга при очаговой ишемии.
Каналы рецепторов ионотропных глутаматов обозначены тремя основными классами, основанными на их преимущественном сродстве к агонистам. Один класс, каналы NMDA-рецепторов, является уникальным, поскольку он лиганд-gated путем одновременного связывания глутаминовой кислоты и глицина и зависит от напряжения. При потенциале покоя мембраны этот канал рецептора блокируется «Mg 2+ plug», даже если оба агониста занимают свои соответствующие сайты связывания. Деполяризация клеточной мембраны удаляет этот блок Mg 2+ и позволяет каналу проводить Na + , K + и Ca 2+
Это дуплексное регулирование является интегральным механизмом в клеточном контроле Ca 2+гомеостаза в нейронах. Ион кальция задействован во множестве молекулярных механизмах, участвующих в различных клеточных процессах, но он также вызывает гибель клеток посредством активации Ca 2+ -зависимых протеаз, образования активных форм кислорода, фосфолипазы A2 и повреждения митохондрий. Напомним читателю, что глутамат является основным нейромедиатором в ЦНС. Каналы NMDA-рецепторов встречаются на большинстве нейронов , где они участвуют в нескольких важнейших аспектах физиологической и патологической активности мозга.
Категория сообщения в блог:
Источник