Механизмы иммунитета система комплемента

Система комплемента, состоящая примерно из 30 белков, как циркулирующих, так и экспрессированных на мембране, является важной эффекторной ветвью как врожденного, так и опосредованного антителами приобретенного иммунного ответов. Термин «комлемент» появился в связи с тем, что этот чувствительный к повышению температуры материал сыворотки крови был обнаружен по свойству «дополнять» способность антител уничтожать бактерии. Известно, что комплемент играет главную роль в защите от многих инфекционных микроорганизмов.
Наиболее важными составляющими его защитной функции являются: 1) выработка опсонинов — молекул, увеличивающих способность макрофагов и нейтрофилов к фагоцитозу; 2) выработка анафилатоксинов — пептидов, индуцирующих местные и системные воспалительные реакции; 3) непосредственный киллинг микроорганизмов.
Известны и другие важные функции комплемента, такие как усиление антигенспецифических иммунных ответов и поддержание гомеостаза (стабильности внутри организма) путем удаления иммунных комплексов и мертвых или умирающих клеток. Мы знаем также, что нарушение контроля над активацией комплемента может привести к повреждению клеток и тканей организма.
Компоненты комплемента синтезируются в печени, а также клетками, участвующими в воспалительной реакции. Концентрация всех белков комплемента в циркулирующей крови составляет примерно 3 мг/мл. (Для сравнения: концентрация IgG в крови составляет примерно 12 мг/мл) Концентрации некоторых компонентов комплемента высоки (например, около 1 мг/мл для С3), в то время как другие компоненты (такие как фактор D и С2) присутствуют в следовых количествах.
Пути активации комплемента
Начальные этапы активации комплемента заключаются в последовательной каскадной активации одного за другим его компонентов. На этой стадии активация одного компонента индуцирует действие фермента, которое приводит к активации следующего по очереди компонента. Поскольку одна активная молекула фермента способна расщеплять множество молекул субстрата, этот каскад реакций усиливает относительно слабый начальный сигнал. Эти каскадные свойства системы комплемента аналогичны наблюдаемым в других сывороточных каскадах, направленных на образование сгустка и выработку кининов, сосудистых медиаторов воспаления.
После активации отдельные компоненты расщепляются на фрагменты, обозначаемые строчными буквами. Меньший из расщепленных фрагментов обычно обозначается буквой «а», больший — «b». Исторически сложилось, однако, что больший из расщепленных фрагментов С2 обычно относят к С2а, а меньший — к С2b. (Однако в некоторых текстах и статьях фрагменты компонентов комплемента С2 обозначаются обратным способом.) Дальнейшие фрагменты расщепления также обозначаются малыми буквами, например C3d.
Известны три пути активации комплемента: классический, лектиновый и альтернативный.
Начало каждого из путей активации характеризуется собственными компонентами и процессами распознавания, однако на более поздних стадиях во всех трех случаях используются одни и те же компоненты. Свойства каждого пути активации и веществ, их активирующих, обсуждаются далее.
Классический путь
Классический путь активации так называется потому, что он был определен первым. Белковые компоненты классического пути обозначаются С1, С2, С9. (Номера расставлены в том порядке, в котором компоненты были открыты, а не в том, в котором они активируются.) Комплексы антиген — антитело являются основными активаторами классического пути. Таким образом, последний является главным эффекторным путем активации гуморального адаптивного иммунного ответа.
Другими активаторами являются некоторые вирусы, погибшие клетки и внутриклеточные мембраны (например, митохондрий), агрегаты иммуноглобулинов и β-амилоид, обнаруживаемый при болезни Альцгеймера в бляшках. С-реактивный белок является белком острой фазы — компонентом воспалительной реакции; он прикрепляется к полисахариду фосфорилхолину, экспрессированному на поверхности многих бактерий (например, Streptococcus pneumoniae), и тоже активирует классический путь.
Классический путь инициируется, когда С1 прикрепляется к антителу в комплексе антиген — антитело, например антителу, связанному с антигеном, экспрессированным на поверхности бактерии (рис. 13.1). Компонент С1 представляет собой комплекс из трех различных белков: Clq (содержащего шесть одинаковых субкомпонентов), связанного с двумя молекулами (причем каждой по две) — Clr и Cls. При активации Cl его глобулярные участки — субкомпоненты Clq — связываются с Clq-специфичным участком на Fc-фрагментах или одного IgM, или двух близко расположенных молекул IgG, связанных с антигеном (связывание IgG показано на рис. 13.1).
Таким образом, антитела IgM и IgG являются эффективными активаторами комплемента. Иммуноглобулины человека, обладающие способностью связываться с Cl и активировать его, в порядке уменьшения этой способности располагаются: IgM > > IgG3 > IgG 1 » IgG2. Иммуноглобулины IgG4, IgD, IgA и IgE не взаимодействуют с Clq не закрепляют и не активируют его, т.е. не активируют комплемент по классическому пути.
После связывания С1 с комплексом антиген—антитело Cls приобретает ферментативную активность. Эта активная форма известна как Cls-эстераза. Она расщепляет следующий компонент классического пути — С4 — на две части: С4а и С4b. Меньшая часть — С4а — остается в растворенном состоянии, а С4b ковалентно связывается с поверхностью бактерии или другой активирующей субстанцией.
Часть С4b, прикрепленная к поверхности клетки, затем связывает С2, который расщепляется Cls. При расщеплении С2 получают фрагмент С2b, который остается в растворенном состоянии, и С2а. В свою очередь С2а прикрепляется к С4b на поверхности клетки с образованием комплекса С4b2а. Этот комплекс называется С3-конвертазой классического пути, поскольку, как мы увидим позднее, этот фермент расщепляет следующий компонент — С3.
Лектиновый путь
Лектиновый путь активируется концевыми остатками маннозы в белках и полисахаридах, находящихся на поверхности бактерии. Эти остатки не обнаруживаются на поверхности клеток млекопитающих, поэтому лектиновый путь может рассматриваться в качестве средства распознавания своего и чужого. Поскольку этот путь активации не требует присутствия антител, он является частью системы врожденной иммунной защиты.
На рис. 13.1 показано, как бактериальные маннозные остатки связываются с циркулирующим комплексом маннозосвязывающего лектина (МСЛ; по структуре схожий с Clq классического пути) и двумя ассоциированными протеазами, называемыми маннозассоциированными сериновыми протеазами (МАСП-1 и -2). Это связывание активирует МАСП-1 для последующего расщепления компонентов классического пути комплемента — С4 и С2 с формированием С4b2а, С3-конвертазы классического пути на поверхности бактерий. А МАСП-2 обладает способностью напрямую расщеплять С3. Таким образом, лектиновый путь после фазы активации С3 аналогичен классическому.
Альтернативный путь
Альтернативный путь активации комплемента запускается почти любой чужеродной субстанцией. К наиболее изученным веществам относятся липополисахариды (ЛПС, также известные как эндотоксины клеточной стенки грамотрицательных бактерий), клеточные стенки некоторых дрожжей и белок, находящийся в яде кобры (фактор яда кобры). Некоторые агенты, активирующие классический путь, — вирусы, агрегаты иммуноглобулинов и мертвые клетки, запускают также и альтернативный путь.
Активация происходит в отсутствие специфических антител. Таким образом, альтернативный путь активации комплемента является эффекторной ветвью системы врожденной иммунной защиты. Некоторые компоненты альтернативного пути характерны только для него (сывороточные факторы В и D и пропердин, известный также как фактор Р), в то время как другие (С3, С3b, С5, С6, С7, С8 и С9) являются общими с классическим путем.
Компонент С3b появляется в крови в небольших количествах после спонтанного расщепления реактивной тиоловой группы в С3. Этот «предсу-ществующий» С3b способен связываться с гидроксильными группами белков и углеводов, экспрессированных на клеточных поверхностях (см. рис. 13.1). Накопление С3b на поверхности клетки инициирует альтернативный путь.
Оно может происходить как на чужеродной, так и на собственной клетке организма; таким образом, с точки зрения альтернативного пути он всегда запущен. Однако, как указано более детально далее, собственные клетки организма регулируют течение реакций альтернативного пути, в то время как чужеродные не обладают такими регуляторными способностями и не могут предотвратить развитие последующих событий альтернативного пути.

Рис. 13.1. Запуск классического, лектинового и альтернативного путей. Демонстрация активации каждого пути и формирования С3-конвертазы
На следующей стадии альтернативного пути сывороточный белок, фактор B, соединяется с С3b на поверхности клетки с формированием комплекса С3bВ. Затем фактор D расщепляет фактор В, который находится на поверхности клетки в комплексе С3bВ, в результате чего образуется фрагмент Ва, который высвобождается в окружающую жидкость, и Вb, остающийся связанным с С3b Этот С3bВb является С3-конвертазой альтернативного пути, которая расщепляет С3 на С3а и С3b.
Обычно С3bВb быстро растворяется, но может стабилизироваться при соединении с пропердином (см. рис. 13.1). В результате стабилизированный пропердином С3bВb способен связываться и расщеплять большое количество С3 за очень короткое время. Накопление на клеточной поверхности этих быстро образованных в большом количестве С3b приводит к почти «взрывному» запуску альтернативного пути. Таким образом, связывание пропердина с С3bВb создает петлю усиления альтернативного пути. Cпособность пропердина активировать петлю усиления контролируется противоположным действием регуляторных белков. Следовательно, активация альтернативного пути не происходит постоянно.
Активация С3 и С5
Расщепление С3 является основной фазой для всех трех путей активации. На рис. 13.2 показано, что С3-конвертазы при классическом и альтернативном путях (С4b2а и С3bВb соответственно) расщепляют С3 на два фрагмента. Более мелкий С3а является растворимым белком анафилатоксином: он активирует клетки, участвующие в реакции воспаления. Больший фрагмент, С3b, продолжает процесс активации каскада комплемента, связываясь с клеточными поверхностями вокруг места активации. Как показано далее, С3b также участвует в защите организма, воспалении и иммунной регуляции.
 и альтернативном (внизу) путях")
Рис. 13.2. Расщепление компонента С3 С3-конвертазой и компонента С5 С5-конвертазой при классическом и лектиновом (наверху) и альтернативном (внизу) путях. Во всех случаях С3 расщепляется на С3b, который откладывается на клеточной поверхности, и СЗа, высвобождаемый в жидкую среду. Таким же образом С5 расщепляется на С5b, который откладывается на клеточной поверхности, и С5а, высвобождаемый в жидкую среду
Связывание С3b с С3-конвертазами как при классическом, так и альтернативном путях инициирует связывание и расщепление следующего компонента — С5 (см. рис. 13.2). По этой причине С3-конвертазы, связанные с С3b, относятся к С5-конвертазам (С4b2а3b при классическом пути; С3bВb3b при альтернативном). При расщеплении С5 образуются два фрагмента. Фрагмент С5а высвобождается в растворимой форме и является активным анафилатоксином. Фрагмент С5b связывается с клеточной поверхностью и формирует ядро для связи с терминальными компонентами комплемента.
Терминальный путь
Терминальные компоненты каскада комплемента — С5b, С6, С7, С8 и С9 — являются общими для всех путей активации. Они связываются друг с другом и формируют мембраноатакующий комплекс (МАК), который вызывает лизис клетки (рис. 13.3).

Рис. 13.3 Формирование мембраноатакующего комплекса. Компоненты комплемента поздней фазы — С5b-С9 — последовательно соединяются и формируют на поверхности клетки комплекс. Многочисленные С9-компоненты прикрепляются к этому комплексу и полимеризуются с образованием поли-С9, создавая канал, который пронизывает клеточную мембрану
Первой фазой формирования МАК является прикрепление С6 к С5b на поверхности клетки. Затем С7 связывается с С5b и С6 и проникает в наружную мембрану клетки. Последующее связывание С8 с С5b67 приводит к образованию комплекса, глубже проникающего в мембрану клетки. На мембране клетки C5b—С8 действует как рецептор для С9 — молекулы типа перфорина, который связывается с С8.
Дополнительные молекулы С9 взаимодействуют в комплексе с молекулой С9, образуя полимеризованные С9 (поли-С9). Эти поли-С9 формируют трансмембранный канал, нарушающий осмотическое равновесие в клетке: через него проникают ионы и поступает вода. Клетка набухает, мембрана становится проницаемой для макромолекул, которые затем покидают клетку. В результате происходит лизис клетки.
Р.Койко, Д.Саншайн, Э.Бенджамини
Опубликовал Константин Моканов
Источник

Система комплемента — комплекс белков, постоянно присутствующих в крови, которые выполняют ряд важнейших функций. На данный момент комплекс насчитывает более трех десятков белковых молекул, среди которых основные белки — С1, С2, …, С9, а также ряд белков-регуляторов. Комплемент принято относить к факторам врожденного иммунитета, выполняющим защитную функцию, однако при определенных обстоятельствах сбой работы системы комплемента может стать звеном патогенеза некоторых заболеваний.
Вначале разберем механизмы работы системы комплемента, после чего рассмотрим их клиническое значение.
Активация системы комплемента
Существует три пути активации данной системы: классический, альтернативный и лектиновый. Все они приводят к ключевому событию: формированию С3-конвертазы.
В классическом варианте комплемент активируется через IgG и IgM, которые формируют иммунные комплексы с антигенами. Комплекс С1 (состоящий из q, r и s субъединиц) связывается с Fc-фрагментом иммуноглобулина. Происходит активация C1, комплекс приобретает протеолитическую активность и активирует C4 и C2. Последние формируют ключевой ферментативный комплекс — С3-конвертазу.
При альтернативном пути активация комплемента происходит без участия антител. Инициируют его поверхностные молекулы микробов и их внеклеточные структуры — например, углеводороды, полисахариды и т. д. С3 в клетках постоянно (даже когда нет активации системы комплемента) претерпевает диссоциацию на С3а и С3b, но в очень малых количествах.
Когда в клетке появляется инициирующий фактор (предположим, это бактерия), С3b связывается с ее поверхностными молекулами. К этому комплексу присоединяются другие молекулы-регуляторы: фактор В, фактор D, пропердин. Так появляется еще одна форма С3-конвертазы.
Лектиновый путь активируется через лектин, связывающий маннозу (фактор врожденного иммунитета — MBL) или группу лектинов Ficolin, которые связывают молекулы на поверхности патогенов — дрожжей, бактерий, паразитов и вирусов. MBL и Ficolin постоянно циркулируют в крови в виде MBL-ассоциированного комплекса (MASP). Когда MASP связывается с вышеназванными молекулами, комплекс претерпевает изменения конформации и активирует уже знакомый путь C4 и C2, что приводит к формированию С3-конвертазы, как и в классическом случае.
Итак, вне зависимости от пути активации различия оканчиваются на формировании ключевого фермента — С3-конвертазы. Последняя расщепляет С3 на два фрагмента: С3а и С3b. Отметим, что С3b выполняет функцию опсонина, С3а — анафилотоксин, способный воздействовать на тучные клетки с высвобождением гистамина.
Однако С3b также способен присоединиться к С3-конвертазе и модифицировать ее, превратив в С5-конвертазу. Этот фермент проделывает то же самое с С5, образуя С5а и С5b. По аналогии, С5а — анафилотоксин, С5b — опсонин.
И снова С5b присоединяется к С5-конвертазе, вовлекая в каскад С6–С9 с формированием т. н. мембраноатакующего комплекса (MAC) [1].
.
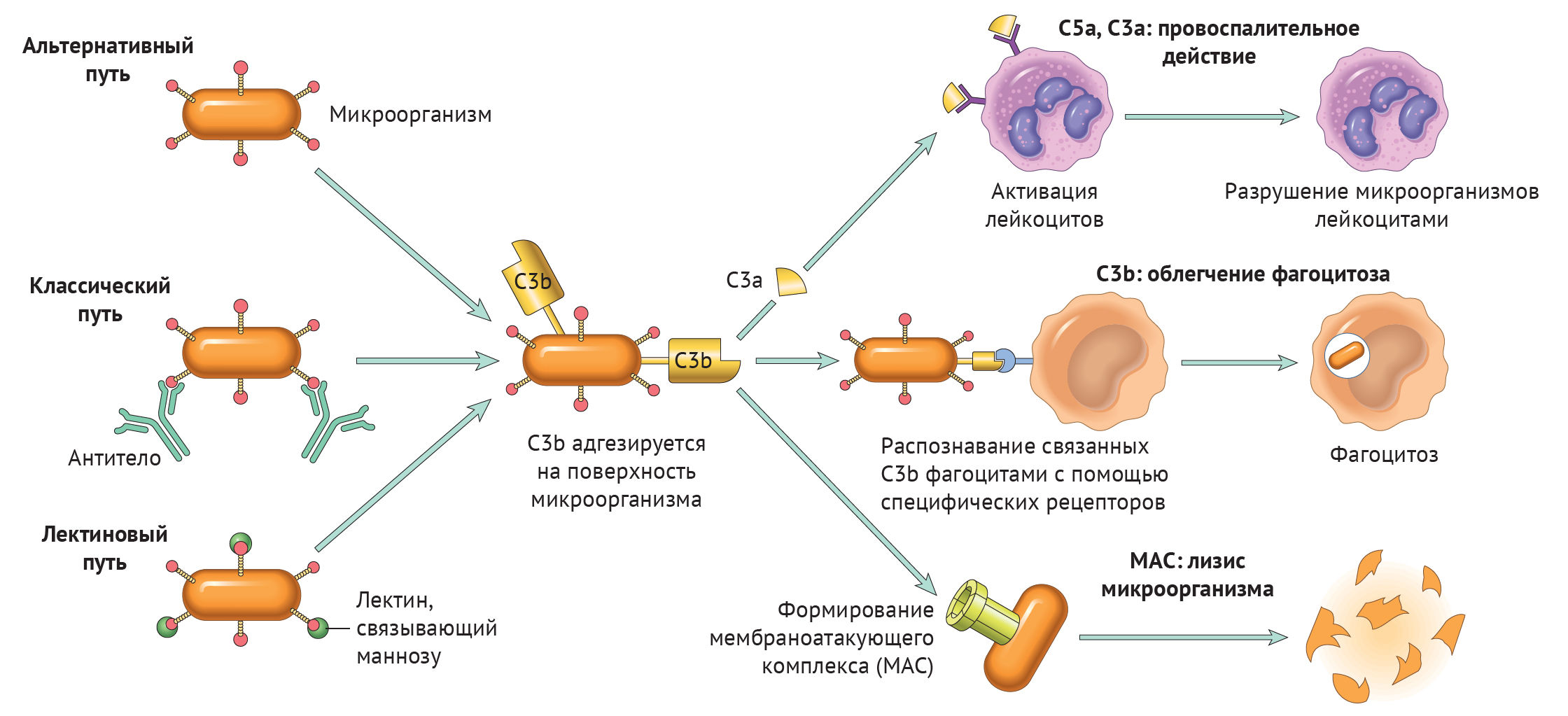
Рисунок 1
Любой из трех путей активации системы комплемента приводит к формированию С3-конвертазы, которая расщепляет С3-компонент на С3а и С3b. Последний участвует в опсонизации патогенов и облегчает таким образом фагоцитоз, а также инициирует каскад формирования МАС. С3а вместе с С5а (и в меньшей степени С4а) — побочные продукты реакций, которые обладают провоспалительным действием («Robbins Basic Pathology», 10nd ed — 2018, p 76).
Таким образом, систему комплемента можно разделить на три функциональные группы: анафилотоксины, опсонины и мембраноатакующий комплекс — МАС.
.
Анафилотоксины
К ним относятся С3а, С4а и С5а. В целом, их роль сводится к провоспалительному действию, что выражается в повышении проницаемости сосудов микроциркуляторного русла (МЦР), вазоконстрикции. В нейтрофилах, эозинофилах и макрофагах анафилотоксины инициируют респираторный взрыв, в базофилах и тучных клетках — высвобождение гистамина. Кроме того, анафилотоксины регулируют синтез эозинофильного катионного белка, адгезию и хемотаксис эозинофилов [2].
В здоровом организме роль анафилотоксинов по большому счету этим и ограничивается. Однако в случае реакций гиперчувствительности данные молекулы могут стать ключевым звеном патогенеза. Одним из наиболее ярких примеров является бронхиальная астма.
Эпителий и гладкомышечные клетки дыхательных путей содержат рецепторы к анафилотоксинам — C3aR и C5aR. При аллергических реакциях происходит активация системы комплемента, который в свою очередь активирует иммунокомпетентные клетки (ИКК), среди которых — нейтрофилы, эозинофилы, макрофаги, дендритные клетки. Последние под влиянием стимула (например, аллергена) способны в свою очередь вновь активировать комплемент — так замыкается порочный круг.
Помимо поддержания системного воспаления, анафилотоксины ответственны и за ремоделирование дыхательных путей. Под их влиянием происходит гиперплазия мерцательного эпителия и гладкомышечных клеток, неоваскуляризация и фиброзирование паренхимы [3,4].
Также анафилотоксины играют важную роль в патогенезе синдрома системного воспалительного ответа (SIRS). При сепсисе, когда микробы в больших количествах попадают в кровоток, происходит системная активация цитокинов, хемокинов и, конечно, системы комплемента. Повышение концентрации анафилотоксинов при сепсисе, к слову, считается неблагоприятным прогностическим фактором [5].
В случае с сепсисом и SIRS анафилотоксины становятся звеном патогенеза. Наибольшая роль в данной ситуации принадлежит С5а. Предположительно, избыточный синтез этого белка приводит к дисфункции нейтрофилов, апоптозу лимфоидных клеток, способствует развитию кардиомиопатий, ДВС-синдрома и полиорганной недостаточности.
В связи с этим постепенно разрабатываются препараты, ингибирующие синтез С5а. На биологических моделях было показано, что блокада данного фактора действительно улучшает прогноз при сепсисе и SIRS [6].
МАС
Данный комплекс может формироваться на поверхности грамотрицательных бактерий и напрямую участвовать в их уничтожении (более всего действие МАС направлено против Neisseria). Описаны также случаи образования МАС на поверхности грамположительных бактерий, паразитов и мембране собственных клеток. Состоит он из последних 5 белков комплемента: С5 — С9.
Специфического рецептора, инициирующего образование комплекса, нет. Белки комплемента адгезируются на наружной поверхности клеточной стенки бактерии, после чего комплекс приобретает ферментативную активность и перфорирует мембрану. Нарушается концентрация ионов и воды — клетка погибает [7].
Недавние исследования показывают, что у МАС имеется и провоспалительное — «сублитическое» — действие. При образовании комплекса на поверхности нейтрофилов или макрофагов происходит локальная утечка провоспалительных медиаторов, а в случае атаки мезангиальных клеток и микроглии — высвобождение цитокинов. Также МАС вызывает образование инфламмасомы путем активации Nod-подобного рецептора [8].
Не допустить образование МАС на собственных клетках помогает рецептор CD59. Генетический дефект CD59 приводит к появлению пароксизмальной ночной гемоглобинурии [7]. А поскольку МАС способен поддерживать системное воспаление, он также вовлекается в патогенез различных воспалительных заболеваний (см. ниже).
Возрастная макулярная дегенерация
Возрастная макулярная дегенерация (ВМД) — основная причина слепоты у пациентов старше 50 лет в цивилизованных странах. В макуле (как и во всей сетчатке) происходят дистрофически-дегенеративные процессы, вследствие чего нарушается способность фокусировать свет в определенной точке, в результате исчезает ясность и четкость зрения. Существует две формы ВМД: влажная (быстро прогрессирующая, экссудативная) и сухая (атрофическая форма). Иногда выделяют рубцовую форму, которая скорее является завершающей стадией заболевания.
Одним из ключевых звеньев этиопатогенеза ВМД является дефект фактора комплемента H — одного из основных регуляторов системы комплемента. Функция его заключается в ингибировании С3-конвертазы (если точнее — является кофактором для протеазы, которая осуществляет катализ С3-конвертазы). Существует несколько форм гена фактора Н, один из наиболее известных — Y402H, который существенно повышает риск развития ВМД. Есть и другие, более редкие варианты [9].
В случае влажной формы ВМД происходит неоваскуляризация сетчатки — аномальное разрастание сосудов под действием VEGF (фактор роста эндотелия сосудов). В опытах было показано, что высвобождение VEGF и неоваскуляризация невозможны без действия MAC [10].
Также в обоих вариантах ВМД важная роль приписывается локальному хроническому воспалению под воздействием системы комплемента. Установлена взаимосвязь между активацией комплемента (особенно по альтернативному пути) и риском развития ВМД [10].
Разумеется, эти данные невозможно было проигнорировать, вследствие чего начали разрабатывать ингибиторы факторов системы комплемента для предотвращения ВМД. На данный момент существуют и моноклональные антитела (анти-фактор D, Экулизумаб), и рекомбинантный фактор Н, и антагонисты различных белков системы комплемента, но пока ни один из них не рекомендован для лечения ВМД [10].
Болезнь Альцгеймера
Воспалительный процесс в нервной ткани сегодня рассматривается как основное патогенетическое звено в развитии БА и других нейродегенеративных заболеваний. Также существуют данные, что прием противовоспалительных препаратов (НПВС) существенно снижает риск развития БА [11].
Однако помимо воспалительных цитокинов, важная роль в развитии локального воспаления принадлежит и системе комплемента. В ликворе пациентов с болезнью Альцгеймера обнаруживается повышенная концентрация С3 в сравнении со здоровыми пациентами. Также активность системы комплемента у пациентов с БА существенно выше [12].
Наследственный ангионевротический отек
НАО — редкое и потенциально фатальное заболевание, этиологическим фактором которого является генетический дефект (НАО 1 типа) или дисфункция (НАО 2 типа) ингибитора С1-компонента комплемента (iС1). В норме iС1 — регулятор, который блокирует весь путь активации системы комплемента. При его дефиците происходит гиперактивация начальных компонентов комплемента, следствием чего является повышение проницаемости сосудов, что приводит к различным отекам.
Проявляется НАО в основном в возрасте после 20 лет отеками конечностей, реже — шеи и лица, которые длятся 1–3, максимум — 7 дней, после чего самостоятельно проходят. Сначала пациент может их даже не замечать, однако со временем частота и выраженность отеков нарастают, может происходить отек слизистой ЖКТ, что проявляется болью, тошнотой, иногда — клиникой острого живота. Наиболее опасен отек гортани, который может сопровождаться асфиксией.
Но ситуация с лечением не так уж и плоха. Ингибитор С1 можно вводить внутривенно для профилактики приступов, для купирования — подкожно [13,14].
Пароксизмальная ночная гемоглобинурия
ПНГ — редкое заболевание, в основе которого лежит клональная экспансия гемопоэтических клеток с мутацией PIGA. Результатом данной мутации является недостаточность ГФИ-заякоренных белков, а именно — CD55 и CD59 (гликозилфосфатидилинозитол или ГФИ-якорь — гликопептид, присоединяющийся к белкам в процессе посттрансляционных модификаций).
Проявляется заболевание гемолитической анемией, костномозговой недостаточностью, тромбофилией.
В норме CD55 ингибирует С9-компонент и предотвращает формирование МАС, а CD59 — ингибирует образование С3-конвертазы и предотвращает протеолиз С3-компонента.
Патогенез начинается с внутрисосудистого гемолиза, который происходит ввиду дефицита CD55. На поверхности эритроцитов с мутацией формируется С3-конвертаза, которая запускает дальнейший синтез МАС. Образование последнего ничего не сдерживает из-за дефекта CD59. МАС перфорирует мембрану эритроцита, и клетка погибает.
Все клинические проявления — гемоглобинурия, анемия (апластическая), тромбоз/эмболия, гастроинтестинальные и неврологические симптомы, так или иначе, являются следствием данного механизма [15,16].
В качестве лечения используется трансплантация гемопоэтических клеток, гемотрансфузии, симптоматическая и поддерживающая терапия. Также достаточно перспективным является использование ингибиторов системы комплемента, например, Экулизумаба (Солириса), который, по данным некоторых публикаций, устраняет все значимые симптомы заболевания [15].
Однако по данным Кохрейна, доказательную базу Экулизумаба нельзя назвать достаточной [17].
Атипичный гемолитико-уремический синдром
Для АГУС характерными симптомами являются гемолитическая анемия, тромбоцитопения и почечная недостаточность с уремией. Этиологией данного заболевания могут быть различные генетические дефекты регуляторов системы комплемента.
Зачастую это мутация CFH, отвечающего за экспрессию уже знакомого читателю фактора Н. Таких мутаций существует более 80, они могут быть наследственными или спорадическими. Также встречаются мутации генов, ответственных за фактор В, С3-компонент комплемента, тромбомодулин и др. [18].
В лечении АГУС также может быть использован экулизумаб [19], однако и здесь ощущается нехватка доказательной базы. В основном же терапия АГУС осуществляется с помощью гемотрансфузий/плазмообмена, диализа, почечной трансплантации — и снова ни одна из названных процедур не способна избавить пациента от болезни [18]. Таким образом, наиболее перспективна разработка ингибиторов системы комплемента и проведение РКИ с их участием.
Помимо перечисленных заболеваний, система комплемента принимает непосредственное участие в патогенезе аутоиммунных кожных заболеваний [20], воспалительных заболеваний почек [21], разнообразных аллергических и аутоиммунных заболеваний.
Разработка ингибиторов комплемента уже сегодня является перспективным направлением, а некоторые препараты данной группы (экулизумаб) уже могут применяться в ряде случаев.
Источники:
- J. V. Sarma and P. A. Ward, ‘The complement system’, Cell Tissue Res., vol. 343, pp. 227–235, 2011.
- J. Kohl, ‘Anaphylatoxins and infectious and non-infectious inflammatory diseases’, Mol. Immunol., vol. 38, no. 2–3, pp. 175–187, 2001.
- M. A. Khan, A. M. Assiri, and D. C. Broering, ‘Complement mediators: Key regulators of airway tissue remodeling in asthma’, J. Transl. Med., vol. 13, no. 1, pp. 1–9, 2015.
- Y. Laumonnier, A. V. Wiese, J. Figge, and C. Karsten, ‘Regulation and function of anaphylatoxins and their receptors in allergic asthma’, Mol. Immunol., vol. 84, pp. 51–56, 2017.
- C. E. Hack et al., ‘Elevated plasma levels of the anaphylatoxins C3a and C4a are associated with a fatal outcome in sepsis’, Am. J. Med., vol. 86, no. 1 C, pp. 20–26, 1989.
- R. S. Hotchkiss, L. L. Moldawer, S. M. Opal, K. Reinhart, I. R. Turnbull, and J.-L. Vincent, ‘Sepsis and septic shock’, Nat. Rev. Dis. Prim., vol. 2, no. 16045, pp. 1–47, 2017.
- C. Bayly-Jones, D. Bubeck, and M. A. Dunstone, ‘The mystery behind membrane insertion: A review of the complement membrane attack complex’, Philos. Trans. R. Soc. B Biol. Sci., vol. 372, no. 1726, 2017.
- B. P. Morgan, ‘The membrane attack complex as an inflammatory trigger’, Immunobiology, vol. 221, no. 6, pp. 747–751, 2016.
- E. C. Schramm, S. J. Clark, M. P. Triebwasser, S. Raychaudhuri, J. M. Seddon, and J. P. Atkinson, ‘Genetic variants in the complement system predisposing to age-related macular degeneration: A review’, Mol. Immunol., vol. 61, no. 2, pp. 118–125, 2014.
- R. Troutbeck, S. Al-Qureshi, and R. H. Guymer, ‘Therapeutic targeting of the complement system in age-related macular degeneration: A review’, Clin. Exp. Ophthalmol., vol. 40, no. 1, pp. 18–26, 2012.
- P. L. McGeer, J. Rogers, and E. G. McGeer, ‘Inflammation, antiinflammatory agents, and Alzheimer’s disease: The last 22 years’, Handb. Infect. Alzheimer’s Dis., vol. 54, pp. 11–15, 2017.
- H. Krance, C. W. Yi, and Z. Huiyan, ‘The complement cascade in Alzheimer ’ s disease : a systematic review and meta-analysis’, Mol. Psychiatry, 2019.
- K. Bork et al., ‘Guideline: Hereditary angioedema due to C1 inhibitor deficiency: S1 Guideline of the German Society for Angioedema (Deutsche Gesellschaft für Angioödeme, DGA), German Society for Internal Medicine (Deutsche Gesellschaft für Innere Medizin, DGIM), German S’, Allergo J. Int., vol. 28, no. 1, pp. 16–29, 2019.
- H. Longhurst et al., ‘Prevention of hereditary angioedema attacks with a subcutaneous C1 inhibitor’, N. Engl. J. Med., vol. 376, no. 12, pp. 1131–1140, 2017.
- A. Kumar, ‘Paroxysmal Nocturnal Hemoglobinuria’, Blood, vol. 124, no. 18, pp. 1462–1470, 2014.
- C. Parker et al., ‘Diagnosis and management of paroxysmal nocturnal hemoglobinuria’, Rev. Transl. Hematol., vol. 106, no. 12, pp. 3699–3709, 2005.
- A. J. Martí-Carvajal, V. Anand, A. F. Cardona, and I. Solà, ‘Eculizumab for treating patients with paroxysmal nocturnal hemoglobinuria’, Cochrane Database Syst. Rev., vol. 2013, no. 2, 2013.
- M. Progress, ‘Atypical Hemolytic–Uremic Syndrome’, N. Engl. J. Med., 2010.
- C. M. Legendre et al., ‘Terminal complement inhibitor eculizumab in atypical hemolytic-uremic syndrome’, N. Engl. J. Med., vol. 368, no. 23, pp. 2169–2181, 2013.
- G. Edwards, G. F. H. Diercks, M. A. J. Seelen, B. Horvath, M. B. A. Van Doorn, and J. Damman, ‘Complement activation in autoimmune bullous dermatoses: A comprehensive review’, Front. Immunol., vol. 10, no. JUN, pp. 1–8, 2019.
- K. Koåcielska-Kasprzak, D. Bartoszek, M. Myszka, M. Åabińska, and M. Klinger, ‘The complement cascade and renal disease’, Arch. Immunol. Ther. Exp. (Warsz)., vol. 62, no. 1, pp. 47–57, 2014.
Источник