На мрт низкое расположение миндалин мозжечка что это такое

Опущение миндалин мозжечка в большое затылочное отверстие или кагал спинного мозга называют дистопией. А иногда эта патология называется аномалией Киари. Как правило, такое заболевание не влечет за собой значительных расстройств или явных симптомов и не имеет причин для беспокойства больного. Часто эта патология проявляется после достижения 30 – 40-летнего возраста. Обнаруживается обычно при обследованиях по другим причинам. Потому об этом заболевании следует знать, чтобы оно не оказалось неожиданностью для больного.
Для того, чтобы понять, что такое низкое расположение миндалин мозжечка, необходимо четко знать клинические симптомы и как обнаруживается патология. А поскольку это состояние сопровождается частыми головными болями, нужно сначала установить их причину, а потом уже заниматься лечением. Такое заболевание выявляется при МРТ.
Причины появления
Дистопия мозжечка, как правило, является врожденной патологией. Она возникает при смещении какого-либо органа в эмбриональный период. Вторичной она бывает лишь при проведении частых пункций или при люмбальных травмах. Иных причин появления данного заболевания не выявлено.
Миндалины мозжечка очень сходны с теми, которые находятся в гортани. В нормальном положении они располагаются выше БЗО черепа. А отклонения в их развитии и положении могут повлечь за собой не только дистопию. Чаще всего встречается опущение миндалин мозжечка ниже уровня черепа.
До сих пор заболевание Киари является патологией, о причинах возникновения которого неврологи не пришли к одному мнению. Некоторые придерживаются мнения, что эта аномалия возникает при уменьшении габаритов ямки позади черепного выходного отверстия к спинному мозговому каналу. Это нередко приводит к таким последствиям в процессе роста тканей, которые расположены в коробке. Они выходят в затылочный выходной канал. Иные специалисты полагают, что заболевание начинает развиваться по причине увеличения объемов мозговых тканей головы. В этом случае мозг начинает выталкивать через заднюю черепную ямку в затылочное черепное отверстие мозжечок и его миндалины.
Вызывает такое прогрессирование выраженной аномалии и ее переход в «клинику», как гидроцефалия. При этом увеличиваются общие размеры мозга, особенно мозжечковые ткани. Патологию Киари вместе с недоразвитым связочным аппаратом мозга сопровождает дисплазия тканей кости. Потому любая черепно-мозговая травма нередко приводит к усилению снижения уровня нахождения миндалин и мозжечка.
Типы аномалий
Есть такие виды аномальных отклонений – дистопия и аномалия Киари.
В свою очередь, заболевание Киари разделяют на четыре различных типа:
- Тип I отличается расположением миндалин ниже уровня большого затылочного отверстия. Определяется такая патология, как правило, у подростков и у взрослых. Часто ее сопровождает скопление спинальной ликворной жидкости в центральном канале, где расположен спинной мозг пациента.
- Тип II характеризуется проявлением сразу после появления на свет. Кроме того, помимо миндалин, при втором типе патологии в отверстие затылочной части выходят червь мозжечка с частью продолговатого мозга и желудочек. Второй тип аномалии много чаще сопровождает гидромиелия, чем при патологии, описанной в первом случае. В большей части случаев такое патологическое отклонение связано с врожденными грыжами, образованными в различных отделах спинного мозга.
- III тип отличают опустившиеся сквозь отверстие не только миндалины, а и мозжечок вместе с тканями продолговатого мозга. Они размещаются в шейном и затылочном отделах.
- Тип IV представляет собой недоразвитие тканей мозжечка. Эта патология не сопровождается смещением их в каудальном направлении. Но при этом аномалию чаще всего сопровождает врожденная киста, располагающаяся в черепной ямке, и гидроцефалия.
II и III типы часто проявляются в сочетании с явлениями дисплазии нервной системы, например, с гетеротопией мозговых тканей коры, кистами отверстия и пр.
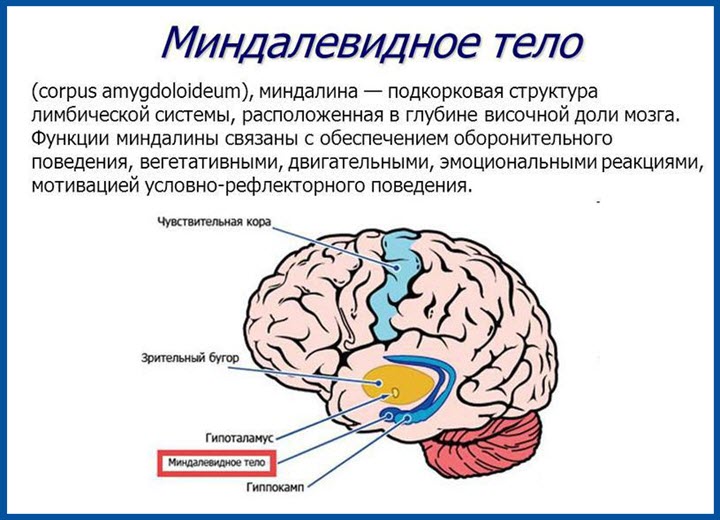
Симптомы
Самой распространенной среди аномалий является патология первого вида. При ней нередко возможно проявление ликворно-гипертензионного синдрома, а также церебелло-бульбарное и сирингомиелическое явления, нарушения работы нервных окончаний внутри черепа.
Ликворно-гипертензионный синдром – это боли в затылке и шейных мышцах, которые усиливаются во время чихания, кашля или напряжения шейных мышечных тканей. Часто боли сопровождаются рвотой, не связанной с приемами еды. Многие симптомы патологии проявляются в зависимости от того, в каком положении располагаются миндалины мозжечка относительно отверстия, которое находится в затылочной ямке черепа. Наблюдается также:
- повышенный тонус мышц шейного отдела;
- нарушения речевых функций;
- ухудшение работы органов зрения и слуха;
- отклонения при глотании;
- частые головокружения, сопровождаемые шумом в голове;
- ощущение вращения окружающей обстановки;
- непродолжительные обмороки;
- перепады давления при резких движениях;
- атрофия языка;
- сиплость голоса;
- нарушения дыхания и чувствительности разных частей тела;
- приступы онемения;
- нарушения в тазовых органах;
- ослабление мышц конечностей.
Аномалия II и III типов имеет похожие симптомы, заметные уже с первых мгновений после рождения малыша. Второй тип сопровождает шумное дыхание, а также неожиданные приступы остановки дыхания, нейропарез гортанных тканей. Наблюдаются также отклонения процесса глотания.
Признаки дистопии редко бывают очевидными. Но все же возможны неврологические проявления:
- приступы «стреляющих» болей в шейных мышцах при усилении напряжения или кашле;
- частые боли в области головы;
- приступы головокружений и обмороки.
Если опущение миндалин сильное, иногда наблюдается расширение канала, связывающего головной и спинной мозг, а также образовываются полости вокруг канала.
Диагностические методы
Основным современным методом диагностирования дистопии является МРТ. В этом случае ни КТ, ни рентгеновские исследования не дают полной картины патологии.
Для диагностирования синдрома Киари не подойдут никакие стандартные методы типа ЭЭГ, ЭхоЭГ или РЭГ, потому как они не позволяют точно поставить диагноз. Осмотр невролога тоже не определит аномалию. Все эти методы могут показать подозрение только на повышенное давление внутри черепной коробки. Рентгенографию черепа также не стоит делать, поскольку она показывает лишь аномалии костных тканей, которые могут сопровождать патологию. Потому до введения в диагностическую практику томографии диагностировать эту болезнь было проблематично. Современные же способы диагностирования позволяют точно определить патологию.
В случае качественной визуализации костных тканей вертебрального перехода такие методы, как МСКТ или КТ, не дают достаточно точной картины. Единственным достоверным способом диагностирования аномалии Киари на сегодня является только МРТ.
Поскольку проведение исследования этим методом требует неподвижности больного, маленьких детей погружают в искусственный сон с помощью лекарств. Проводится также МРТ спинного мозга. Она направлена на диагностику любых аномальных отклонений в работе нервной системы.
Лечение
Консервативные методы лечения возможны лишь при очень незначительных отклонениях. Все зависит от того, каково состояние больного на момент обращения к врачу. В этом случае лечение направлено на снятие болезненных симптомов нестероидными лекарствами или миорелаксантами. Необходима также коррекция режима.
Единственным эффективным методом лечения при обширных отклонениях является хирургическое вмешательство, которое заключается в расширении ямки черепа и пластики твердых мозговых тканей оболочки.
Показаниями для оперативного лечения являются:
- изнуряющие головные боли, которые не снимаются препаратами.
- нарастание мозговых проявлений, приводящее к инвалидности.
Если аномальное отклонение протекает без каких-то ощутимых признаков, лечения не требуется. В случаях возникновения болезненных ощущений в районе шеи и затылка проводится консервативная терапия, при которой используют анальгетики и асептические лекарственные вещества, а также миорелаксанты.
При сопровождении аномалии Киари нарушениями неврологических функций или когда консервативный курс терапии не дает результатов, назначается хирургическая операция.
Часто при лечебных курсах синдрома Киари используется метод краниовертебральной декомпрессии. Операция подразумевает расширение отверстия затылочной части за счет удаления части костной ткани, отсечения мозжечковых миндалин и части двух позвонков шеи. Благодаря этому, нормализуется оборот цереброспинальной жидкости в мозговых тканях в результате выполнения заплаты из аллотрансплантата или искусственного материала. Иногда синдром Киари лечат с помощью шунтирования, которое позволяет дренировать цереброспинальную жидкость из центрального канала. Посредством хирургической операции можно отвести цереброспинальную жидкость в сосуды органов груди или брюшины.
Прогноз
Аномалия Киари первого типа может протекать бессимптомно всю жизнь. И третий тип патологии почти всегда приводит к смертельному исходу, если не осуществить своевременное лечение. В случае появления неврологических признаков заболевания первого или последнего типов очень важно своевременно проведенное хирургическое лечение, потому как появившийся недостаток неврологических функций будет плохо восстанавливаться, даже если успешно провести манипуляции. По разным данным, результативность хирургической операции отмечается примерно в половине эпизодов.
Источник
Синдром Арнольда-Киари

Во время формирования плода могут возникнуть различные аномалии, в числе которых – синдром Арнольда-Киари или мальформация Арнольда-Киари.
При этом отклонении головной мозг еще до рождения сдавливается из-за того, что отдел черепа, где находится мозжечок или малый мозг, как его еще называют, чересчур мал или деформирован.
В результате происходит смещение миндалин плода в верхний отдел позвоночного канала с их последующим ущемлением. Малый объем задней доли черепа приводит к тому, что у взрослых развивается синдром Арнольда-Киари 1 типа.
Причины возникновения патологии
Четкие причины возникновения синдрома Арнольда-Киари не в полной мере выяснены учеными. Это не связано с хромосомными аномалиями, по крайней мере, такая связь пока не выявлена.
Есть две точки зрения на этот вопрос: ранее считалось, что мальформация Арнольда-Киари является врожденной, а в последнее время ученые предполагают, что этот дефект – приобретенный.
Процент пациентов с врожденной аномалией Арнольда-Киари гораздо меньше, чем процент людей, которые этот синдром приобрели.
Врожденные причины
- Кости черепа у плода развиваются не так, как нужно: слишком маленькая задняя черепная ямка, где у нормально формирующегося плода находится мозжечок. Таким образом, синдром Арнольда-Киари возникает в результате несоответствия роста мозга и черепных костей, то есть кости плода «не успевают» за его мозгом.
- Гиперувеличение большого затылочного отверстия у плода.
Приобретенные причины
- Появление черепно-мозговых травм у плода во время родов.
- Повреждение особой жидкостью (ликвором) у плода спинного мозга. В результате растягивается его центральный канал.
Кроме того, синдром Арнольда-Киари может возникнуть одновременно или вследствие иных пороков, например, водянки спинного мозга.
Факторы риска
Достаточно велика вероятность того, что риск развития у плода аномалии Арнольда-Киари увеличивают следующие факторы:
- злоупотребление лекарствами при беременности, особенно если их не выписывал врач;
- курение, употребление алкоголя во время вынашивания плода;
- перенесение матерью плода вирусных заболеваний, например, краснухи.
Типы мальформации Арнольда-Киари
Все типы синдрома Арнольда-Киари связаны с патологически низким расположением миндалин мозжечка. Однако они отличаются друг от друга структурой ткани головного мозга, которая попадает в позвоночный канал, наличием патологии развития головного или спинного мозга и степенью вклинивания продолговатого мозга в позвоночник.
Назовем типы мальформации Арнольда-Киари:
- 1 тип – низко, в шейном отделе, располагаются миндалины мозжечка, однако другие отклонения от нормы в головном мозге отсутствуют.
- 2 тип указывает на смещение мозжечка в большое затылочное отверстие, сочетающееся с иными аномалиями головного мозга, позвоночника и спинного мозга.
- 3 тип – появление грыжи в затылке и полное смещение структур заднего мозга в расширенное затылочное отверстие. С таким типом заболевания человек не сможет долго прожить, этот этап чреват летальным исходом.
Симптомы
1 и 2 типы синдрома Арнольда-Киари гораздо более распространены, чем 3 тип мальформации. Но это совершенно разные болезни, поэтому рассмотрим симптомы каждой из них в отдельности.
Симптомы синдрома Арнольда-Киари 1 и 2 типа
Самый отличительный симптом аномалии Киари 1 типа – перманентная головная боль. Кроме нее для данного этапа заболевания характерны:
- тошнота, рвота;
- руки утрачивают чувствительность, становятся слабыми;
- сильно болит шея;
- шум в ушах;
- походка становится неуверенной;
- в глазах двоится;
- глотание затруднено, речь становится смазанной.
Симптомы синдрома Арнольда-Киари 2 типа развиваются обычно сразу же после рождения или в раннем детском возрасте. Перечислим их:
- проблемы с глотанием – верный симптом аномалии такого заболевания 2 типа.
- нарушение дыхания, слабый крик, свистящее шумное дыхание у новорожденных с аномалией Киари.
Симптомы в более тяжелых случаях аномалии:
При более тяжелой степени синдрома Арнольда-Киари могут наблюдаться состояния, угрожающие инфарктом головного и спинного мозга. Перечислим эти симптомы:
- если больной поворачивает голову, у него временно может появиться слепота или двоение в глазах, даже потеря сознания;
- дрожание конечностей, нарушение координации;
- пол-лица, часть туловища, рука или нога могут частично потерять чувствительность;
- мышцы частей лица, туловища, рук, ног ослабевают;
- различные проблемы с мочеиспусканием.
Соответственно и методы лечения различных типов мальформации тоже отличаются.
Лечение синдрома Арнольда-Киари
Существует несколько методов лечения мальформации Арнольда-Киари: хирургическое вмешательство и применение нехирургических методов. Выбор того или иного пути лечения зависит от симптомов, связанных с синдромом Арнольда-Киари, или их отсутствия, от степени тяжести каждого конкретного пациента. Так, лечение будет зависеть именно от проявленных симптомов заболевания.
Консервативное (нехирургическое) лечение
При слабовыраженной симптоматике пациент просто регулярно подвергается профилактическим осмотрам. При незначительной боли в шейно-затылочной области врач назначает обезболивающие, противовоспалительные средства, препараты, расслабляющие мускулатуру.
Таким больным нужно делать физические упражнения, может быть полезна трудотерапия, все, что поспособствует улучшению мышечной координации и устранит дрожь. Возможны занятия с логопедом.
Все эти меры могут отсрочить или полностью предотвратить проведение нижеследующего способа лечения.
Хирургическое лечение
Если применение обычных методов лечения синдрома Арнольда-Киари не приносит желанного эффекта или больного серьезно беспокоят такие симптомы, как онемение конечностей, слабость в них, расстройство зрения, потеря сознания, то обязателен оперативный метод лечения мальформации.
Нейрохирургическая операция производится, во-первых, с целью исправления каких-либо дефектов, которые приводят к сдавливанию головного мозга, во-вторых, ради приведения в норму движения ликвора. В результате оперативного вмешательства при лечении вышеуказанной болезни приостанавливается прогрессирование мальформации в структуре позвоночника и мозга, стабилизируются симптомы.
Синдром Арнольда-Киари 1 типа лечится путем избавления от небольшого фрагмента кости, хирургически удаленного из заднего отдела черепной ямки. Это довольно распространенная операция при таком типе синдрома, которая длится пару часов, а восстанавливается больной в течение недели.
В результате проведения операции увеличивается пространство для мозга и уменьшается внутричерепное давление. Синдром Арнольда-Киари 2 типа лечится с помощью шунтирования и закрытия миеломенингоцеле.
Успешный исход операции определяется уменьшением давления на спинной мозг и мозжечок, нормализацией оттока ликвора.
Профилактика синдрома Арнольда-Киари
Профилактика – лучшее лечение, но, к сожалению, на 100% действенные способы предотвратить синдром Арнольда-Киари пока науке неизвестны. Это связано с неполными сведениями о причинах возникновения аномалии.
Единственное, что обязательно должны делать родители будущего ребенка, особенно мать, вынашивающая плод, это по максимуму устранить перечисленные выше факторы риска, влияющие на развитие мальформации Арнольда-Киари.
Кроме того, женщина, вынашивающая плод, должна следовать правилам здорового образа жизни:
- полноценно питаться;
- не курить и не пить алкоголь, не говоря уже о наркотиках;
- принимать лекарства только по назначению врача.
Источник: https://prosindrom.com/neurological/sindrom-arnolda-kiari.html
Арнольда-Киари Аномалия I

Аномалия Арнольда-Киари I – опущение миндалин мозжечка в большое затылочное отверстие со сдавливанием продолговатого мозга. Может сочетаться с сирингомиелией, базилярной импрессией или инвагинацией, ассимиляцией атланта. Симптомы коррелируют от степени снижения. Методом выбора в диагностике является магнитно-резонансная томография (МРТ).
Оперативное лечение, ламинэктомия (удаление дужек верхних шейных позвонков) с декомпрессивной краниоэктомией задней черепной ямки и пластикой твёрдой мозговой оболочки, применяется только при наличии у пациента неврологического дефицита с отсутствием эффекта от консервативной терапии в течение 2-3 месяцев.
авторы Dr Henry Knipe and Dr Frank Gaillard et al.
Эпидемиология
Аномалия Арнольда-Киари I встречается чаще у женщин [2].
Клиническая картина
В отличие от пороков развития Киари II, III и IV, аномалия Арнольда Киари I часто остается бессимптомной. Вероятность стать клинических проявлений пропорциональна степени опускания миндалин.
Все пациенты с пролабированием миндалин больше 12 мм имеют какую-либо симптоматику, тогда как примерно в 30% пролабирование в диапазоне между 5 и 10 мм протекают бессимптомными [1].
Компрессия ствола мозга (продолговатого мозга) может вызывать сирингомиелию с соответствующими симптомами и клинической картиной (затылочные боли, нарушение глотания, атаксия) разной выраженности, симптомами поражения спинного мозга и др.
Сопутствующие заболевания
Сирингомиелия шейного отдела позвоночника встречается в ~35% (варьирует от 20 до 56%), гидроцефалиия в 30% [1,3] случаев, в обоих случаях считается данные изменения развиваются в результате нарушения ликовродинамики, центральном канале и вокруг спинного мозга.
В ~35% (23-45%) выявляются скелетные аномалии [1, 3]:
- платибазия / базилярная импрессия
- атланто-затылочная ассимиляция
- деформация Шпренгеля (Sprengel)
- синдром Клиппель-Фейля (Klippel-Feil)
Патология
Аномалия Арнольда Киари I характеризуется пролабированием миндалин мозжечка через большое затылочное отверстие, в основном в результате несоответствия между размерами мозжечка и задней черепной ямке.
Аномалию Арнольда Киари I следует отличать от эктопии миндалин, которая протекает бессимптомно и является случайной находкой, при которой, миндалины выступают через затылочное отверстие не более чем на 3-5 мм [1-2].
Рентгенологические особенности
Патология выявляется путем измерения максимального расстояния на которое миндалины выступают ниже плоскости большого затылочного отверстия (условной линии между ophisthion и basion), значения используемые для постановки диагноза отличаются у разных авторов [2]:
- выше затылочного отверстия: норма
- 6 мм: аномалия Арнольда Киари 1
Некоторые авторы используют более простую градацию [1]:
- выше затылочного отверстия: норма
- 5 мм: аномалия Арнольда Киари 1
Положение миндалин мозжечка меняется с возрастом. У новорожденных миндалины расположены чуть ниже большого затылочного отверстия и спускаются ниже с ростом ребенка, достигая своей низшей точки в возрасте 5 – 15 лет.
В дальнейшем они поднимаются на уровень большого затылочного отверстия [3].
Таким образом, снижение миндалин на 5 мм у ребенка будет скорее всего нормой, а взрослом возрасте данные изменения следует рассматривать с подозрением [3].
КТ
Современное объемное сканирование с высоким качеством сагиттальной реформации относительно хорошо визуализирует затылочное отверстие и миндалины, хотя отсутствие контрастности (по сравнению с МРТ) с трудом позволяет провести точную оценку.
Чаще патология может быть заподозрена на аксиальных изображениях, когда мозговое вещество охватывает миндалины а спинномозговая жидкость представлена в малом количестве или отсутствует.
Данное состояние называется, в переполнением затылочного отверстия.
МРТ
МРТ исследование является методом выбора. Сагиттальные срезы наиболее оптимальны для оценки аномалии Арнольда Киари I. Осевые изображения так же дают картину «переполненого» затылочного отверстия.
Лечение и прогноз
Аномалии Арнольда Киари I можно разделить на три этапа (хотя мало данное деление практически не используется в повседневной практике):
- бессимптомная
- компрессия ствола мозга
- сирингомиелия
Оперативное лечение применяется только при наличии у пациента неврологического дефицита с отсутствием эффекта от консервативной терапии в течение 2-3 месяцев. Оно состоит в ламинэктомии (удаление дужек верхних шейных позвонков) с декомпрессивной краниоэктомией задней черепной ямки и пластикой твёрдой мозговой оболочки.
История
Впервые была описана в 1891 году Хансом Киари, Австрийским патологоанатом (1851-1916).
Дифференциальная диагностика
дифференциальный ряд включает в себя:
- эктопия миндалин:
Источник: https://radiographia.info/article/anomaliya-arnolda-kiari-i
Источник