Симптом глаза тигра на мрт

Нейродегенерация с отложением железа в мозге или Болезнь Галлервордена — Шпатца — очень редкое нейродегенеративное заболевание, сопровождающееся отложением железа в базальных ганглиях (в бледном шаре и в чёрной субстанции). Это аутосомно-рециссивно наследуемое заболевание было впервые описано в 1922 году Юлиусом Галлеворденом и Гуго Шпатцем. Частота заболевания в среднем 1-3 человека на 1 миллион.
Патогенез[править | править код]
У большинства пациентов, особенно при ранней манифестации заболевания, определяются мутации в кодируемом фермент пантотенкиназу (PANK2) гене, в хромосоме 20p13. Этот фермент играет решающую роль в биосинтезе кофермента-А. Дефект фермента ведет к накоплению цистеина, который в присутствии железа (что означает — в особенности в области чёрного вещества и базальных ганглиев) ведет к повышению свободных радикалов и способствует оксидатному повреждению мозга. Процесс в целом приводит к отложению железа и нейромеланина.
Протекание заболевания[править | править код]
Обычное начало заболевания в детском возрасте, иногда с выраженной симптоматикой уже на первом году жизни. Редко возможна манифестация во взрослом возрасте. Сначала возникают экстрапирамидные моторные нарушения, в особенности нарушения ходьбы с тенденцией к падениям или дистонией ног, реже — психические нарушения. В дальнейшем — нарушения движений (дистонии, хореоатетоз, тремор) с ригидностью затылочной мускулатуры, гиперрефлексией и нарушениями психики (как правило, расстройство интеллекта в виде прогрессирующей деменции). Также обнаруживаются дизартрия и дисфагия. Можно обнаружить пигментацию сетчатки или атрофию зрительного нерва[2]. У взрослых преобладает синдром паркинсонизма-плюс — с деменцией, гиперрефлексией и проминентной дистонией.
Течение заболевания прогрессивно, неврологическое состояние пациентов ухудшается постепенно.
Смерть наступает обычно в возрасте до 30 лет, в состоянии полного психического распада личности.
Дополнительная диагностика[править | править код]
Необходимо биохимическое исключение болезни Вильсона-Коновалова, факультативно — исключение нейроакантоцитоза, прежде всего с помощью МРТ. В МРТ в Т2- взвешенных изображениях являются типичными — обусловленные отложением железа — гипоинтенсивные очаги в бледном шаре, с центральным очагом гиперинтенсивности — так называемые «глаза тигра». Этот симптом обнаруживается у всех больных с PANK2-мутациями. В генетическом обследовании могут обнаруживаться мутации в PANK2-гене. Тем не менее, уверенно можно говорить о диагнозе только после патологоанатомического исследования.
Лечение[править | править код]
Каузальная (этиологическая) терапия неизвестна. Были попытки лечения энзимного дефекта. Хелаторы («ловушки») железа, такие как Дефероксамин, не оказывают эффекта, тем не менее, с 2007 года проводятся попытки проводить лечение хелатором железа Феррипрокс (Деферипрон®). В экспериментах на животных глубокая стимуляция мозга приводила к усилению дистоний и гиперкинезов. Гипокинезия может лечиться Леводопой, гиперкинезы — антихолинэргиками. Тем не менее, эффект Леводопы у пациентов с мутацией гена PANK2 очень сомнителен.
Для мышечной релаксации и купирования болевого синдрома часто назначается Баклофен или бензодиазепины.
Название заболевания[править | править код]
Из-за связей Юлиуса Галлервордена с программой эвтаназии в фашистской Германии было предложено называть заболевание «нейродегенерация с отложением железа в мозге 1» (англ. neurodegeneration with brain iron accumulation 1). В мире распространено сокращённое название заболевания как «NBIA1».
Примечания[править | править код]
Литература[править | править код]
- Hochspringen ↑ Zhou u.a.: A novel pantothenate kinase gene (PANK2) is defective in Hallervorden-Spatz syndrome. in: Nature genetics (Nat Genet.). New York 28.2001,4 (Aug), 345—349. PMID 11479594 ISSN 1061-4036
- Hochspringen ↑ PKAN-Artikel bei genereviews von Gregory/Hayflick, revidierte Version Jan. 2008
- Hochspringen ↑ M. Shevell: Hallervorden and history. in: The New England journal of medicine (N Engl J Med.). Waltham Mass 2.2003,1 (Jan),348,3-4. PMID 12510036 ISSN 0028-4793
Источник

Болезнь Галлервордена-Шпатца — нейродегенеративная наследственная патология, обусловленная отложением железа в базальных ганглиях головного мозга. Проявляется синдромом паркинсонизма, нарушениями интеллектуальной сферы и психики, гиперкинезами, зрительными расстройствами. Основное диагностическое значение имеет обнаружение рисунка «глаз тигра» в зоне бледного шара при проведении МРТ церебральных структур. Лечение симптоматическое: агонисты дофамина, вальпроаты, антиконвульсанты, нейролептики, антидепрессанты. Прогноз неблагоприятный.
Общие сведения
Болезнь Галлервордена-Шпатца описана в 1922 г. немецкими морфологами, в честь которых и получила свое название. К наиболее типичным клиническим маркерам данной патологии относят гиперкинезы, синдром паркинсонизма, интеллектуальное снижение, атрофию зрительных нервов, пигментную ретинопатию. Болезнь Галлервордена-Шпатца встречается крайне редко. В зависимости от времени ее манифестации различают детскую, ювенильную (подростковую) и взрослую формы. Ранее заболевание диагностировалось лишь посмертно по данным аутопсии. После внедрения в практическую неврологию МРТ стала возможна прижизненная постановка диагноза. Прорыв в изучении этиологии был сделан в 2001 г., когда было установлено, что в основе заболевания лежит генетический дефект, обуславливающий нарушения в синтезе фермента пантотенаткиназы. После этого болезнь Галлервордена-Шпатца была официально переименована в пантотенаткиназа-ассоциированную нейродегенерацию.

Болезнь Галлервордена-Шпатца
Причины болезни Галлервордена-Шпатца
Болезнь Галлервордена-Шпатца является генетической патологией, носящей как семейный, так и спорадический характер, передающейся по наследству аутосомно-рецессивным путем. Генетическим субстратом выступают аберрации в гене пантотенаткиназы (локус 20р12.3–р13 20-й хромосомы). Всего известно более 50 мутаций. Результатом генетического дефекта является уменьшение продукции пантотенаткиназы, что ведет к аккумуляции в базальных структурах цистеина. Последний образует устойчивые химические соединения с ионами железа, которые неблагоприятно воздействуют на белки и запускают процесс перекисного окисления, приводящий к апоптозу нейронов. На месте некротизированных нейронов происходит разрастание глиальной ткани.
Описанный патологический процесс затрагивает преимущественно бледный шар и черное вещество (субстанцию nigra), где морфологически обнаруживаются внеклеточные отложения железа, имеющие коричневую пигментацию. Кроме этого, имеют место сфероидные периаксональные образования, расположенные в белом церебральном веществе, коре мозга, спинном мозге и периферических нервных стволах.
Симптомы болезни Галлервордена-Шпатца
Классическим вариантом болезни Галлервордена-Шпатца считается ранняя детская форма с клинической манифестацией в период от 4 до 10 лет (обычно после 5-летнего возраста). В 90% случаев первым признаком заболевания выступает торсионная дистония, затрагивающая мышцы ног. Ведущей жалобой является затруднение ходьбы. Затем, как правило, происходит генерализация процесса с его распространением на мышцы глотки, лица, туловища. Наряду с генерализованными вариантами могут отмечаться мультифокальный или сегментарный тип дистонии. Наиболее часто наблюдается писчий спазм, блефароспазм, лицевой параспазм, спастическая кривошея. У трети пациентов отмечаются признаки паркинсонизма: мышечная ригидность и гипокинезия. В ряде случаев имеют место эпилептические приступы.
Болезнь Галлервордена-Шпатца характеризуется когнитивными расстройствами в виде снижения внимательности и памяти с постепенным развитием олигофрении; психическими изменениями с преобладанием агрессивности и асоциального поведения. Отмечается дизартрия. У большинства больных имеются нарушения остроты зрения. В 68% случаев они обусловлены атрофией зрительных нервов, в 29% случаев — пигментной ретинопатией. Для детской формы болезни Галлервордена-Шпатца типично быстрое прогрессирование с полной потерей в течение 10-15 лет способности к передвижению.
Подростковый вариант болезни Галлервордена-Шпатца проявляется в возрасте от 10 до 18 лет и характеризуется более замедленным течением. Дебютирует проявлениями фокальной торсионной дистонии, наиболее часто в мышцах конечностей или ортомандибулярной области. Сопровождается психическими, интеллектуальными и поведенческими расстройствами.
Взрослая форма болезни Галлервордена-Шпатца дебютирует после 18-летнего возраста. У большинства больных протекает в виде синдрома паркинсонизма. Характерно обеднение активных движений (гипокинезия), генерализованная ригидность мышц (симптом «зубчатого колеса»), постуральный тремор и неустойчивость (атаксия). Оценка последней возможна при помощи пробы Тавенарда — попыток вывести пациента из состояния равновесия в положении стоя, путем подталкивания его за плечи вперед и в стороны. Главной особенностью является сочетание паркинсонизма с различными гиперкинезами (миоклониями, фокальной торсионной дистонией, атетозом, гемибализмом). Степень нарушения когнитивных функций может варьировать от полной сохранности до прогрессирующей деменции. Возможны эмоциональная лабильность, депрессия, агрессивность, эпиприступы.
Диагностика болезни Галлервордена-Шпатца
Благодаря полиморфизму симптоматики, постановка диагноза болезни Галлервордена-Шпатца представляет трудную задачу для неврологов. Основными критериями заболевания считаются дебют в возрасте до 30 лет, экстрапирамидные расстройства, неуклонное прогрессирование симптомов, наличие типичной МРТ-картины. К дополнительным признакам отнесены наличие пирамидных знаков, прогрессирующее интеллектуальное снижение, эпиприступы, атрофия зрительных нервов, пигментная атрофия сетчатки, аутосомное наследование по рецессивному типу.
В диагностике опираются на данные неврологического статуса и электроэнцефалографии. При нарушении зрения проводят консультацию офтальмолога, визиометрию, офтальмоскопию. Определение типа наследования осуществляет генетик путем составления генеалогического древа. Возможна ДНК-диагностика (поиск мутаций в гене пантотенаткиназы). При проведении ПЭТ головного мозга удается выявить сниженный метаболизм в зоне паллидума. Основанием для исключения болезни Галлервордена-Шпатца является наличие симптомов другой патологии, в рамки которой может укладываться имеющаяся клиническая картина: болезни Вильсона, хореи Гентингтона, нейроакантоцитоза, болезни Мачадо-Джозефа.
Основополагающим методом диагностики болезни Галлервордена-Шпатца выступает МРТ. Во всех типичных вариантах патологии в режиме Т2 на МРТ головного мозга определяется расположенная в области бледного шара гиперинтенсивная зона овальной формы, окруженная еще большей гипоинтенсивной зоной. Подобная МРТ-картина является патогномоничной и получила название «глаз тигра». Гипоинтенсивная зона представляет собой «глаз», а гиперинтенсивная — его «зрачок». Время появления этого признака на томограммах пока дискутируется. По мнению одних авторов «глаз тигра» может появляться еще до клинической манифестации болезни, по мнению других — через несколько лет от дебюта клинических симптомов.
Лечение и прогноз болезни Галлервордена-Шпатца
В настоящее время болезнь Галлервордена-Шпатца не имеет эффективных методов лечения. Попытки терапии препятствующими накоплению железа хелатными соединениями (дефероксамином) и антиоксидантами не имели успеха. В связи с этим применяется симптоматическое лечение. Синдром паркинсонизма служит показанием к назначению дофаминовых агонистов (пирибедила, прамипексола) или производных амантадина. Однако при данном заболевании он, как правило, резистентен к проводимому лечению.
При гиперкинезах применяют вальпроаты, бензодиазепины (диазепам, клоназепам). При спастике рекомендованы миорелаксанты (баклофен, толперизона гидрохлорид), при эпиприступах — топирамат или вальпроаты, при когнитивных расстройствах — ипидакрин и холина альфосцерат, при психических отклонениях — нейролептики (рисперидон, кветиапин, клоназепам), антидепрессанты 3-го поколения (венлафаксин, циталопрам, дапоксетин).
Симптоматическая терапия болезни Галлервордена-Шпатца позволяет уменьшить проявленность клинических симптомов, продлить способность пациентов к самообслуживанию. Вместе с тем продолжается разработка новых способов лечения. Исследуется эффективность применения пантотеновой кислоты. Получены данные о положительном влиянии на течение заболевания магнитной стимуляции бледного шара.
Прогноз зависит от формы заболевания. Наиболее неблагоприятное течение имеет ранняя форма, при которой полная инвалидизация наступает в промежутке от 10 до 15 лет с момента дебюта симптомов. Более благоприятен взрослый вариант, особенно в случаях, когда деменция слабо выражена. Его средняя продолжительность составляет более 20 лет.
Источник
Зоопарк в голове
«Загляни глубже в природу, и ты поймешь все лучше.»
Альберт Эйнштейн
Изображения, получаемые при нейровизуализации, иногда вызывают ассоциации, например, с животными. Такие ассоциации легче запоминаются клиницистами в качестве маркеров различных нейродегенеративных заболеваний. Некоторые из них прошли испытание временем и стали общепризнанными. Другие ждут своей очереди быть описанными находчивыми врачами и стать представителями неврологического «зоопарка».
«Глаз тигра»
«Глаз тигра» впервые описан Sethi et al. в 1988 у двух пациентов с болезнью Галлервордена-Шпатца. Заболевание относится к группе наследственных нейродегенеративных заболеваний с аутосомно-рецессивным типом наследования. У большинства больных выявляется мутация гена пантотенаткиназы 2-го типа, приводящая к нарушению обмена железа. Поражаются преимущественно базальные ганглии головного мозга. При классической форме заболевание дебютирует в детском возрасте (средний возраст дебюта – 3 года) с дистонии конечностей или цервикальной дистонией с последующей генерализацией. Помимо дистонии, клиническими проявлениями могут быть спастические парезы, тремор, миоклония, хорея, эпиприпадки, паркинсонизм, когнитивные нарушения, пигментная ретинопатия и атрофия зрительного нерва. Через 5-10 лет пациенты теряют возможность к самостоятельному передвижению, появляются нарушения глотания и дыхания, что вскоре приводит к смерти. Клиника атипичных вариантов гетерогенна. При частичном снижении активности пантотенкиназы наблюдается дебют заболевания в подростковом или даже взрослом возрасте, течение заболевания более медленное. В клинической картине может доминировать паркинсонизм. Часто он сочетается оромандибулярной дистонией, дизартрией и нейропсихиатрическими нарушениями. Способность к самостоятельному передвижению утрачивается через 15-40 лет.
Формирование «глаза тигра» на изображениях МРТ головного мозга в Т2 режиме связано с поражением бледного шара. В центре отмечается повышение сигнала в связи с гибелью нейронов, глиозом. Вокруг формируется зона пониженного сигнала, что связано с патологическим накоплением железа. У небольшой части больных отмечается лишь снижение интенсивности сигнала от бледного шара. «Глаз тигра» считается патогномоничным признаком болезни Галлервордена-Шпатца, он присутствует более, чем в 95% случаев. Однако он может выявляться и при некоторых других заболеваниях с нарушением обмена железа и у клинически здоровых людей. (рисунок 1)
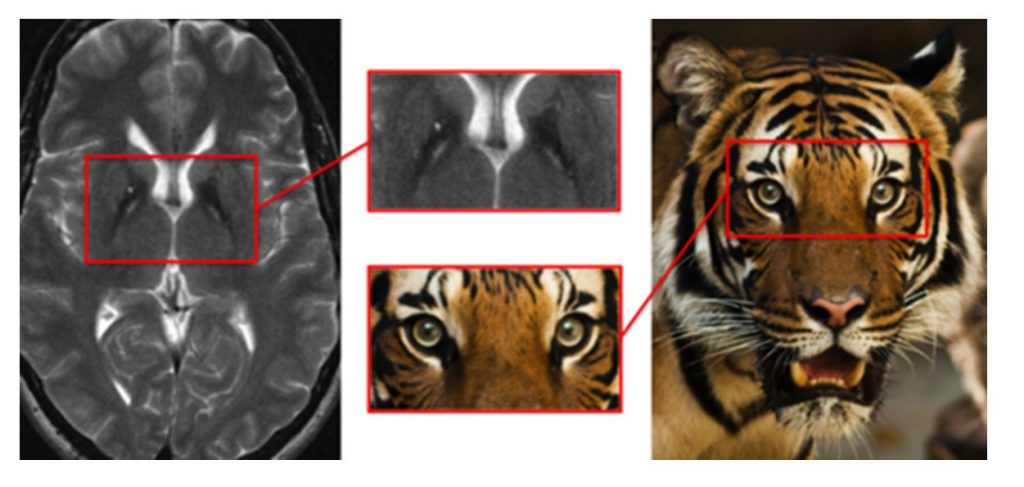
Рисунок 1. «Глаз тигра» при болезни Галлервордена-Шпатца.
«Мордочка панды»
Гепатолентикулярная дегенерация (болезнь Вильсона-Коновалова) – аутосомно-рецессивное заболевание, развивающееся в результате мутации в гене ATP7B. В основе заболевания лежит нарушение элиминации меди. Излишек меди накапливается в печени, головном мозге, роговице, почках и других органах. При дебюте в детском и подростковом возрасте болезнь чаще начинается с симптомов поражения печени, исходом которого может быть цирроз. В возрасте старше 20 лет болезнь чаще начинается с неврологических проявлений: тремора, дистонии, дизартрии, хореоатетоза, паркинсонизма, атаксии, аффективных и когнитивных нарушений. Гепатолентикулярную дегенерацию необходимо исключать у каждого больного с экстрапирамидным синдромом, проявившемся в возрасте до 50 лет.
В случае развития неврологической симптоматики при МРТ головного мозга всегда выявляются отклонения, такие как повышение сигнала в режиме Т2 от базальных ганглиев, таламуса, ствола и белого вещества полушарий. «Мордочку гигантской панды» можно увидеть на аксиальных срезах на уровне среднего мозга: гиперинтенсивный сигнал от покрышки, красных ядер и ретикулярной части черной субстанции, гипоинтенсивный сигнал от впроводящих путей. «Мордочку детеныша панды» можно увидеть на аксиальном срезе на уровне моста: гипоинтенсивный сигнал от покрышки и медиального продольного пучка («глаза»), верхних ножек мозжечка («ушки») и гиперинтенсивный сигнал вокруг водопровода («рот и нос») (рисунок 2).
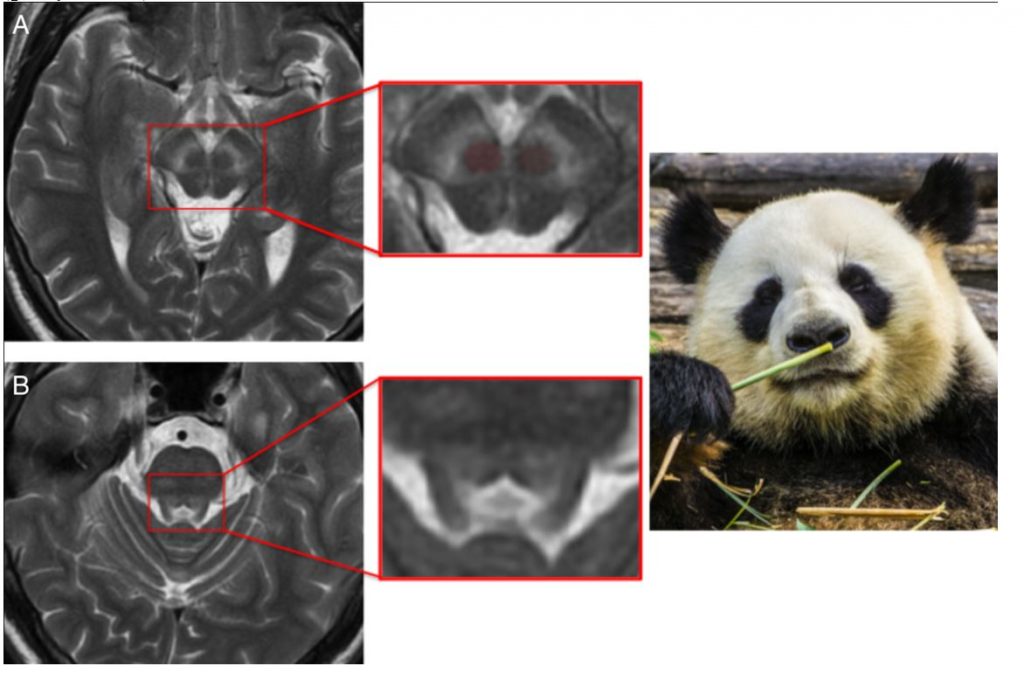
Рисунок 2. «Мордочка панды» при болезни Вильсона-Коновалова.
«Хвост ласточки»
В патогенезе развития болезни Паркинсона большую роль играет поражение черной субстанции, особенно ее дорсолатеральной части. В норме при МРТ головного мозга в режиме ДВИ определяется гиперинтенсивность в каудальных и медиолатеральных отделах черной субстанции в виде «хвоста ласточки». К моменту клинического дебюта болезни Паркинсона наблюдается гибель 60-80% дофаминергических нейронов компактной части черной субстанции, что при нейровизуализации отражается как исчезновение «хвоста ласточки». Однако, данный симптом часто наблюдается и в случаях атипичного паркинсонизма и не позволяет проводить дифференциальную диагностику болезни Паркинсона и заболеваний, протекающих с синдромом паркинсонизма. (рисунок 3)
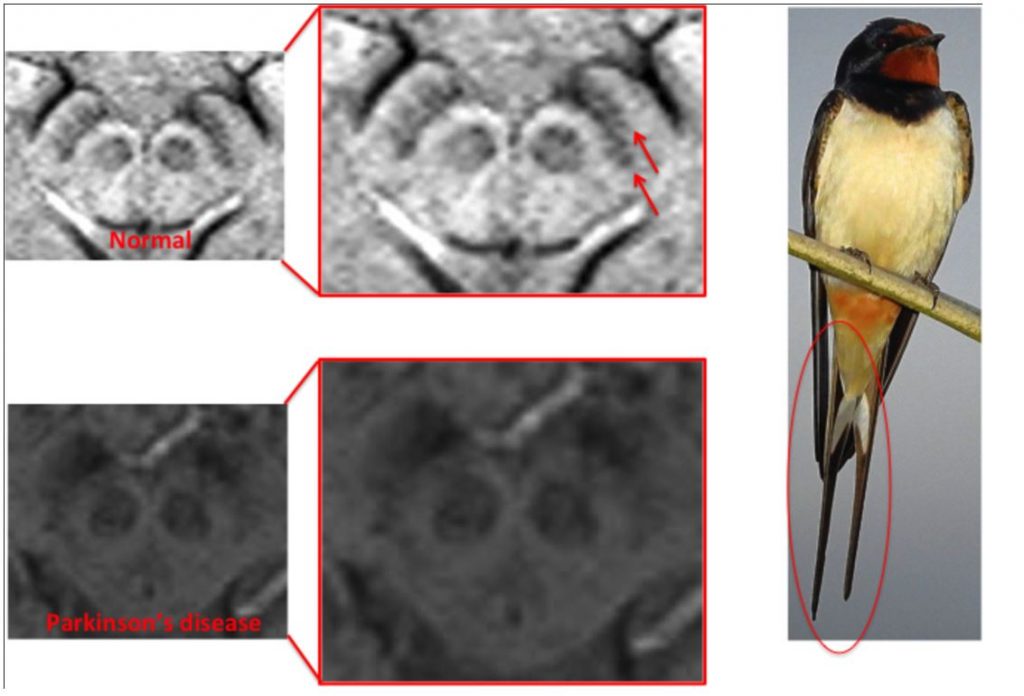
Рисунок 3. «Хвост ласточки» при паркинсонизме.
Симптомы колибри или королевского пингвина
и симптом Микки-Мауса.
Симптом колибри (или симптом королевского пингвина) можно выявить при нейровизуализации у пациентов с прогрессирующим надъядерным параличом. Клинически заболевание проявляется аксиальной ригидностью, псевдобульбарным синдромом, ранним развитием постуральной неустойчивости, когнитивным снижением надъядерным параличом вертикального взора. Симптом колибри отражает избирательную атрофию покрышки среднего мозга, затрагивающую ростральную часть интерстициального ядра медиального продольного пучка, с поражением которого связывают развитие паралича вертикального взора. При МРТ головного мозга на сагиттальном срезе видно пролабирование вниз крыши среднего мозга, что формирует очертания верхней поверхности головы и клюва «колибри» или «пингвина». Симптом может не выявляться на ранних стадиях заболевания, но является высокоспецифичным. Описаны случаи выявления «колибри» при нормотензивной гидроцефалии и сидроме ломкой Х-хромосомы. (рисунок 4)
Рисунок 4. Симптом колибри при прогрессирующем надъядерном параличе.
Симптом Микки-Мауса можно выявить также при прогрессирующем надъядерном параличе, но уже на аксиальных срезах среднего мозга. Его атрофия приводит к «похуданию щечек мышонка». (рисунок 5)
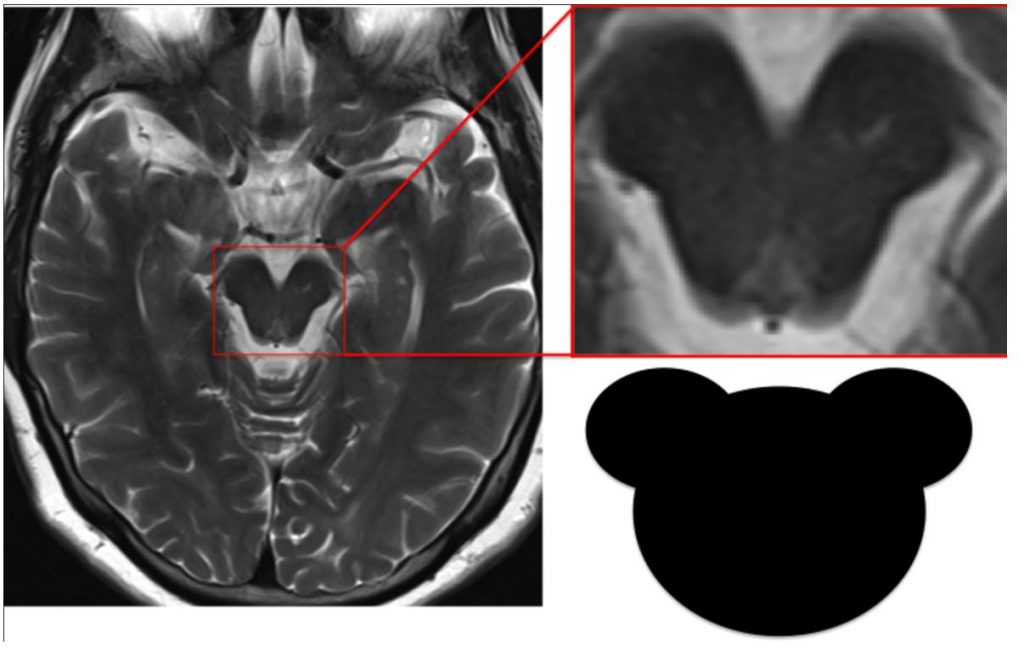
Рисунок 5. Симптом Микки-Мауса при прогрессирующем надъядерном параличе.
«Уши рыси»
Группа наследственных спастических параплегий включает большое количество заболеваний. Наследственные параплегии 11 и 15 типов клинически проявляются прогрессирующим спастическим нижним парапарезом в сочетании с когнитивными нарушениями и синдромом поражения двигательного нейрона. У части пациентов в дебюте развивается паркинсонизм или дистония. Начало заболевания как правило приходится на первое десятилетие жизни. При МРТ головного мозга выявляется истончение мозолистого тела. В режимах Т2 и FLAIR выявляется гиперинтенсивный сигнал от белого вещества около передних рогов боковых желудочков по форме напоминающий кисточки на ушах рыси. Похожие изменения могут выявляться при болезни Маркиафавы-Биньями, после коллозотомии или сочетаться с аномалиями развития мозолистого тела. (рисунок 6).
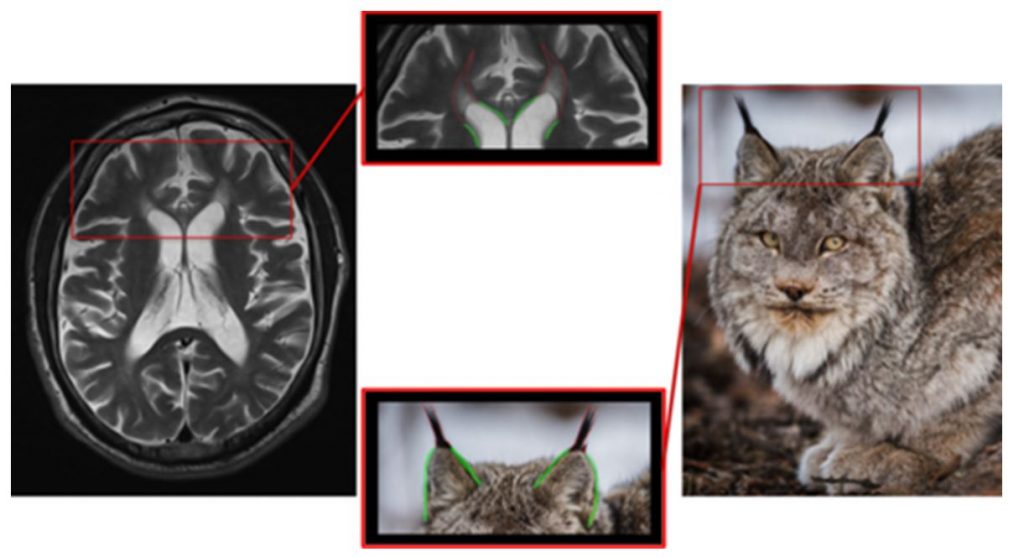
Рисунок 6. Уши рыси при наследственных спастических параплегиях.
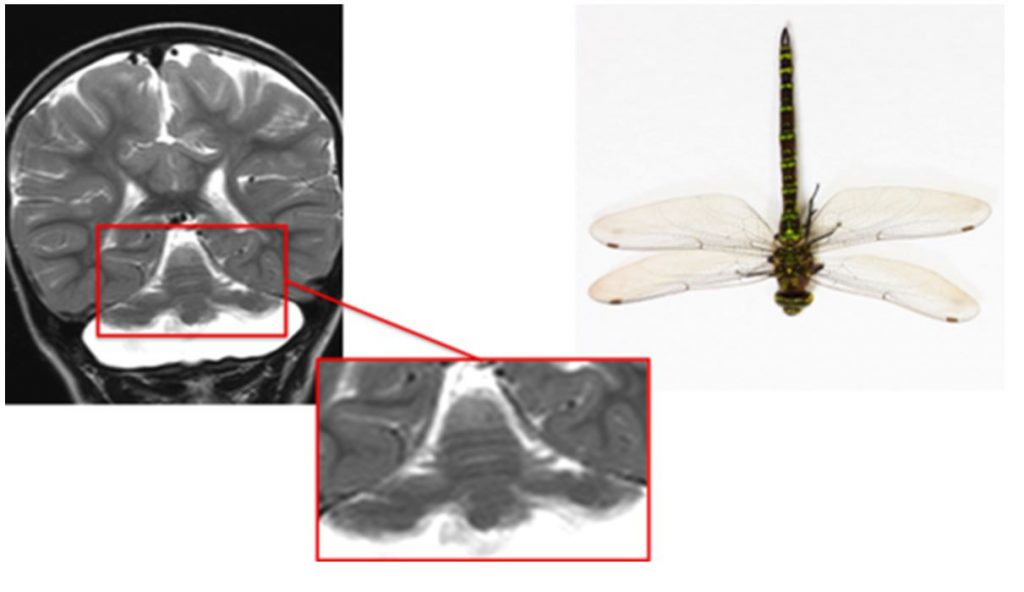
Симптом стрекозы
Понтоцеребеллярная гипоплазия 2 типа – редкое аутосомно-рецессивное нейродегенеративное заболевание. Заболевание манифестирует в первые месяцы жизни – появляются трудности при кормлении, отставание в развитии, заторможенность. Типичны экстрапирамидные нарушения в виде хореоатетоза и дистонии. Пациентам с этим заболеванием часто ошибочно выставляется диагноз ДЦП. (рисунок 7)
Симптом головастика
Болезнь Александера – наследственное демиелинизирующее заболевание с аутосомно-доминантным типом наследования. Классический вариант заболевания проявляется на первом году жизни, но возможен дебют и во взрослом возрасте. Клинически проявляется симптомами поражения ствола головного мозга, мозжечка и спинного мозга. Симптом головастика можно увидеть на сагиттальных срезах головного мозга вследствие выраженной атрофии продолговатого мозга и шейного отдела спинного мозга («хвостик головастика») при сохранности моста. Кроме симптома головастика, можно выявить изменение интенсивности сигнала от средних ножек мозжечка в режиме FLAIR. (рисунок 8)
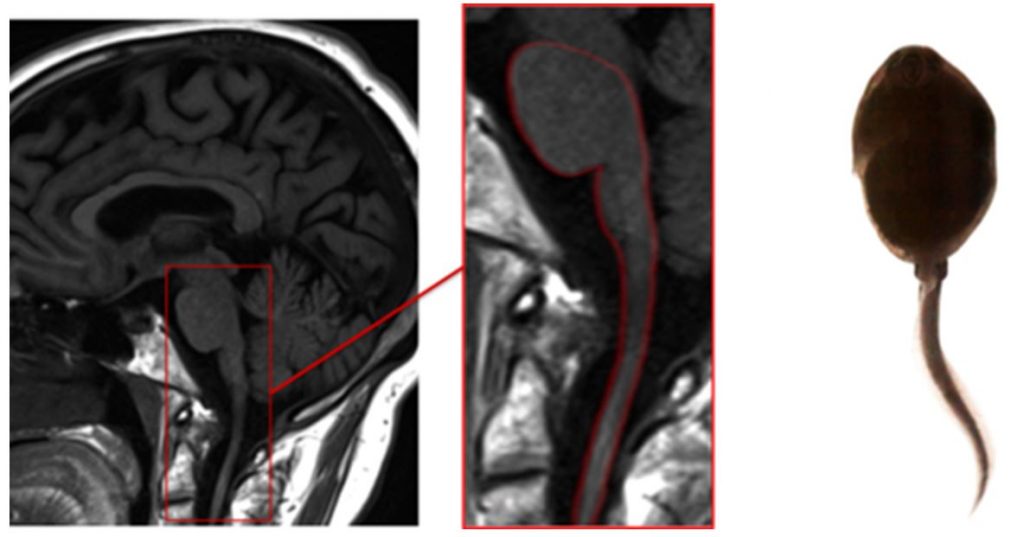
Рисунок 8. Симптом головастика при болезни Александера.
Симптом шкуры тигра или леопарда
Метахроматическая лейкодистрофия – наследственный липидоз с аутосомно-рецессивным типом наследования. В зависимости от возраста дебюта выделяют детскую, ювенильную и взрослую формы. Клинические проявления включают когнитивное снижение, атаксию и периферическую нейропатию. Изменения на МРТ головного мозга выявляются рано и коррелируют с выраженностью клинических проявлений. В режиме Т2 выявляется гиперинтенсивность мозолистого тела и перивентрикулярного белого вещества в лобной и теменной областях. Симптом шкуры тигра можно увидеть из-за чередования полос с нормальной интенсивностью сигнала и гиперинтенсивных. Феномен может наблюдаться и при других лизосомальных заболеваниях, при болезни Пелицеуса-Мерцбахера. (рисунок 9)
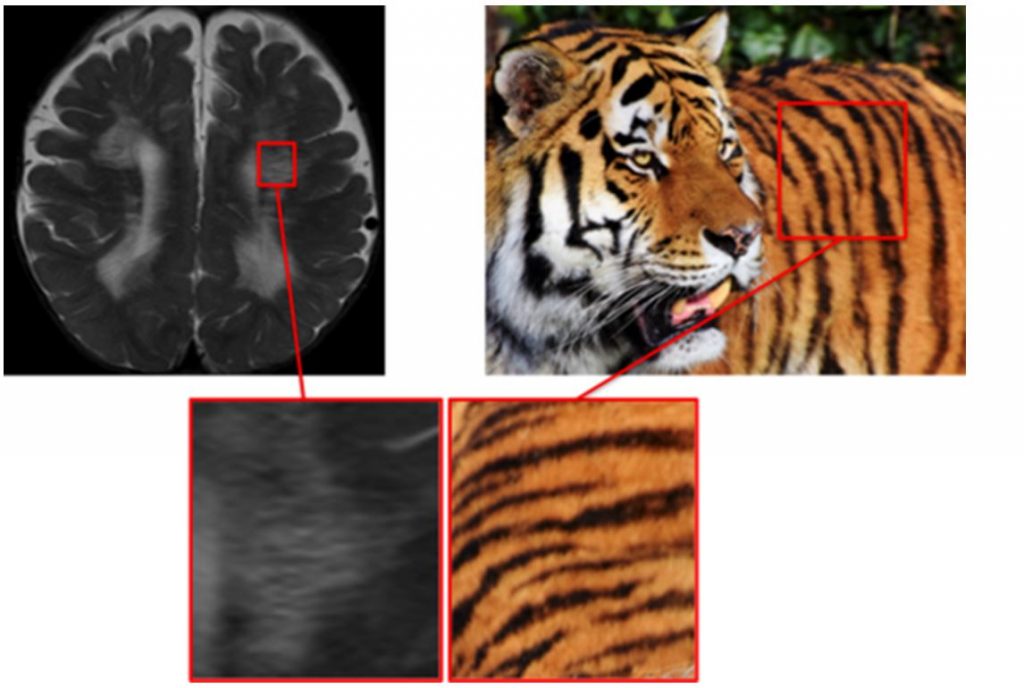
Рисунок 9. Симптом шкуры тигра при метахроматической лейкодистрофии.
Симптом крыла летучей мыши
Глутаровая ацидурия 1 типа – редкое аутосомно-рецессивное заболевание. Развивается вследствие нарушения метаболизма лизина и триптофана. Накопление токсичных продуктов метаболизма приводит к поражению стриатума с развитием дистонии. При МРТ головного мозга выявляется выраженная атрофия с значительным расширением Сильвиевой борозды в виде крыльев летучей мыши. Во многих странах на это заболевание проводится скрининг новорожденных. Строгая диета предотвращает развитие неврологической симптоматики.
Синдром Жубер – наследственное заболевание с манифестацией в неонатальном периоде. Клинически проявляется гипотонией, нарушениями дыхания, окуломоторной апраксией. Позднее присоединяются атаксия, когнитивные и двигательные нарушения. У пациентов, доживающих до взрослого возраста, могут развиться проявления паркинсонизма. При нейровизуализации выявляется аплазия червя мозжечка, удлинение верхних мозжечковых ножек в виде «симптома большого коренного зуба» и расширение четвертого желудочка в форме крыла летучей мыши. (рисунок 10)
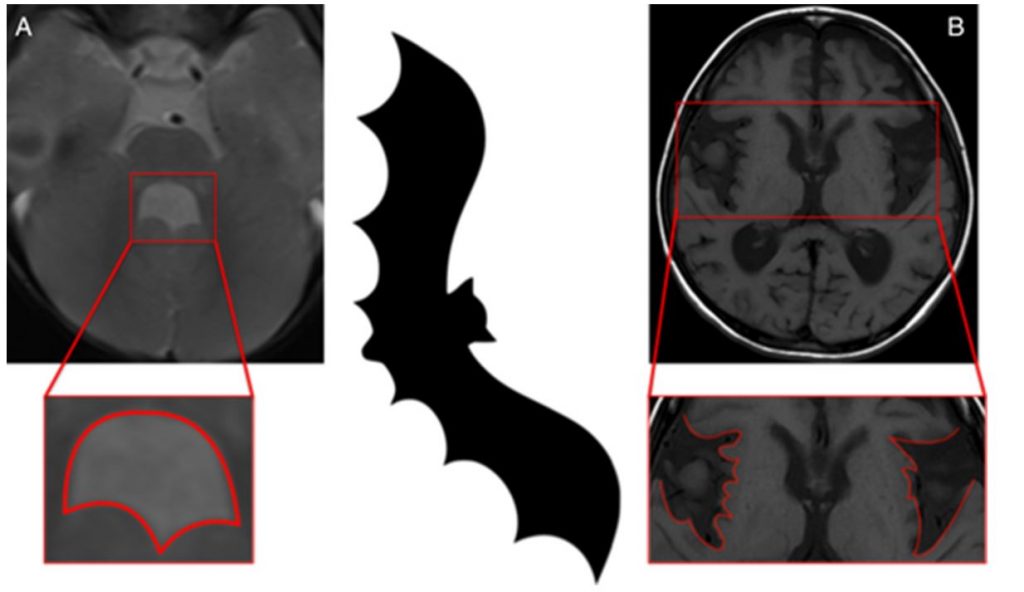
Рисунок 10. Четвертый желудочек в форме крыла летучей мыши при синдроме Жубер. Симптом крыльев летучей мыши при глутаровой ацидурии 1 типа.
Автор: Чеботарева А.Д.
Источник