Симптом глаза змеи на мрт

[читать] (или скачать)
статью в формате PDF
Болезнь Хираямы [БХ] является редким типом шейной миелопатии, характеризующейся преимущественно односторонним или двусторонним асимметричным поражением верхних конечностей, развитием патологии в юношеском возрасте. Данное заболевание принято связывать с аномальным смещением кпереди задней стенки дурального мешка, приводящим к компрессии нижней части шейного отдела спинного мозга, что реализуется хронической ишемией серого вещества спинного мозга.
С момента первого описания БХ в 1959 г. констатировано около 200 клинических ее наблюдений (большую часть больных составляют жители Японии и стран Азии (Китай, Индия), однако существует ряд описаний клинических случаев БХ и из других стран) с использованием различных терминов для данной патологии: ювенильная спинальная мышечная атрофия, ювенильная асимметричная сегментарная мышечная атрофия, ювенильная мышечная атрофия дистальных отделов верхней конечности, ювенильная флексионная шейная мономиелитическая амиотрофия и др. Наиболее часто в публикациях используется прежнее название данной патологии — БХ.
Механизм и причины, приводящие к развитию БХ, по-прежнему остаются дискутабельными. Однако большинство исследователей сходятся во мнении о приоритетности несбалансированности роста, приводящего к диспропорциональности между длиной позвоночного столба и содержимым спинномозгового канала, что влечет за собой узость дурального мешка. Известно, что в норме твердая мозговая оболочка неплотно прилежит к спинному мозгу и имеет места прикрепления в области большого затылочного отверстия (БЗО), С2, С3 и далее у копчика, а также плотно прилежит к позвоночному каналу в месте выхода спинномозговых корешков. У здоровых лиц твердая мозговая оболочка имеет своего рода «люфт» и так называемые поперечные складки, позволяющие компенсировать увеличение длины шейного отдела позвоночника (ШОП) при сгибании. У пациентов же с БХ укороченная твердая мозговая оболочка не может скомпенсировать увеличение ее длины при флексии, что ведет к смещению последней кпереди и последующую компрессию спинного мозга. По мнению ряда авторов, укорочение дурального мешка усиливается в рамках бурного роста в пубертатном периоде. Данный факт объясняет преимущественность манифестации клинических проявлений в юном возрасте, а также приоритетность поражения мужчин относительно женщин (7 : 1).
Существует мнение о том, что одним из механизмов развития БХ служат утолщение и изменение количества эластических волокон твердой мозговой оболочки, в результате снижается ее эластичность и как следствие возникает относительное укорочение, что в дальнейшем ведет к схожим динамическим нарушениям, описанным выше. Другие исследователи предполагают, что у пациентов с БХ отсутствие задних цервикальных эпидуральных связок влечет за собой отсутствие связей между желтой связкой и задней стенкой твердой мозговой оболочки, в результате последняя смещается кпереди при сгибании ШОП, повреждая спинной мозг.
Независимо от причин переднее смещение задней стенки твердой мозговой оболочки ведет к компрессии спинного мозга и, возможно, к хронической ишемии передних отделов серого вещества спинного мозга вследствие повторяющейся компрессии передней спинномозговой артерии. Существует несколько описаний клинических семейных случаев развития со схожей симптоматикой, позволивших предположить, что данное заболевание ‒ есть проявление атопического миелита, вызванного повышенным уровнем иммуноглобулина Е и наличием мутации супероксиддисмутазы-1.
Диагностика БХ основывается на особенностях [1] клинической картины, [2] течения заболевания и [3] дополняется специфичными для данной патологии МРТ-данными. Диагностическими критериями служат:
1 — дистальное поражение преимущественно верхней конечности — снижение силы и атрофия мышц;
2 — возраст начала заболевания от 10 до 20 — лет (15 — 38 лет);
3 — одностороннее или преимущественно одностороннее проявление симптоматики;
4 — характерное начало с постепенным прогрессированием симптомов на протяжении первых нескольких лет заболевания, сменяющееся стабилизацией клинических проявлений и отсутствием прогрессирования;
5 — отсутствие значимых чувствительных нарушений и изменения проприорефлексов, а также симптомов поражения нижних конечностей;
6 — исключение других причин поражения спинного мозга (сирингомиелия, спинальные опухоли, болезнь мотонейрона и т.д.).
В настоящее время считается, что к БХ следует относить только лишь те клинические случаи, которые отвечают приведенным выше диагностическим критериям в дополнении с характерными для данной нозологии особенностями МРТ-картины.
увеличить МРТ-скан №1
увеличить МРТ-скан №2
увеличить МРТ-скан №3
увеличить МРТ-скан №4
Наиболее частыми МРТ-находками при БХ являются изменение нормальной кривизны ШОП, уменьшение площади соприкосновения между задней стенкой дурального мешка и телом соответствующего подлежащего позвонка, локализованная атрофия нижней части шейного отдела спинного мозга, асимметричное уплощение спинного мозга и гиперинтенсивный сигнал в веществе спинного мозга на Т2-взвешанных изображениях некомпрессионного генеза. Данный сигнал, расположенный в передних отделах спинного мозга, имеет на аксиальных срезах характерный вид («симптом глаз змеи»), но не является строго специфичным для мономиелитической миелопатии и может быть также при других шейных миелопатиях. По мнению разных авторов, при мономиелитической амиотрофии он выявляется в 18 — 44% случаев.
Одним из достоверных критериев для постановки диагноза БХ служит обнаружение на аксиальных снимках разобщенности задней стенки твердой мозговой оболочки с подлежащей вертебральной пластиной на протяжении более одной трети от длины последней. Смена лордоза путем выпрямления или кифозирования ШОП является неспецифической, но частой находкой у пациентов с БХ. По мнению ряда авторов, ШОП бывает выпрямленным или кифозированным при приближении или пересечении линии, проведенной от задней поверхности тела С3 к телу С6 позвонка, с линей, соединяющей задние поверхности тел С2 и С7 позвонков, которые при нормальной физиологической кривизне ШОП (лордозе) разобщены между собой.
МРТ в положении флексии демонстрирует смещение кпереди твердой мозговой оболочки, влекущее за собой расширение эпидурального пространства. Данное обстоятельство на аксиальных снимках можно наблюдать в виде гиперинтенсивного в режимах Т1 и Т2 сигнала в форме полумесяца. Подобного рода исследования показывают, что увеличение заднего субарахноидального пространства сочетается с компрессионным уплощением спинного мозга и наличием гиперинтенсивного в Т2 режиме сигнала в передних его отделах.
СМОТРЕТЬ послойные МРТ-сканы при БХ на RADIOPAEDIA.org
Однако исследования последних лет показали относительность указанных выше (1 – 6) критериев. Одним из важных параметров диагностики БХ является дебют заболевания в молодом возрасте. Однако в последние годы в литературе появляются описания клинических случаев развития заболевания у лиц за пределами ювенильной группы. Так, дебют БХ был отмечен у больных в возрасте 41 года и 50 лет. Позднее начало заболевания объясняется возможностью его существования у них в более мягких формах в молодом возрасте с последующим прогрессированием симптомов в течение жизни. Принято считать, что у большинства пациентов наблюдается односторонняя амиотрофия, в некоторых случаях ‒ асимметричная двусторонняя симптоматика, в более редких ‒ двусторонняя.
Лечение больных с БХ сводится к ношению воротника Шанца (для ограничения сгибания шеи), причем это следует начать на самых ранних этапах заболевания и при минимальных МРТ изменениях спинного мозга. Обсуждаются возможности хирургического лечения (пластика твердой мозговой оболочки, передняя декомпрессия шейного отдела спинного мозга). Тщательный отбор пациентов для хирургического лечения должен быть основан на индивидуальном подходе. Рассматривается возможность использования оперативного лечения только для наиболее тяжелых и быстро прогрессирующих случаев. Прогноз при болезни Хираяма благоприятный.
Несмотря на изучение БХ на протяжении более полувека, он продолжает оставаться объектом пристального внимания со стороны исследователей в связи с появлением все более новых фенотипов, не всегда укладывающихся в соответствующую классическую клинико-нейровизуальную картину.
Диагноз БХ сложен и может быть поставлен в результате длительного динамического наблюдения за пациентом с учетом данных клинико-неврологического, нейрофизиологического и нейровизуализационного обследования. При клиническом синдроме нижнего мотонейрона практикующий невролог должен проводить тщательное обследование и дифференциальный диагноз, учитывая при этом возможность доброкачественного течения заболевания, не исключая дебют бокового амиотрофического склероза.
Литература:
статья «Болезнь Хираяма. Описание клинического случая и обзор литературы» Е.Р. Баранцевич, Д.И. Руденко, О.В. Посохина, А.А. Яковлев, Р.А. Гапешин, А.Г. Смочилин, Е.О. Щербакова; ФГБОУ ВО «Первый Санкт-Петербургский государственный медицинский университет имени академика И.П. Павлова» МЗ РФ; ФГБУ «Северо-Западный федеральный медицинский исследовательский центр им. В.А. Алмазова» Министерства здравоохранения Российской Федерации, Санкт-Петербург; ФГБОУ ВО «Санкт-Петербургский государственный университет» МЗ РФ (журнал «Ученые записки СПбГМУ им. акад. И.П. Павлова» №1, 2017) [читать];
статья «Мономелическая амиотрофия (болезнь Хираяма): клинико-лучевые сопоставления» Ж.И. Савинцева, Н.А. Тотолян, Т.Н. Трофимова, Т.Ю. Скворцова, Л.Н. Прахова, А.Г. Ильвес; Институт мозга человека им. Н.П. Бехтеревой Российской академии наук, Санкт-Петербург, Россия; Клиника «Скандинавия», ООО «Ава-Петер», Санкт-Петербург, Россия; Первый Санкт-Петербургский государственный медицинский университет им. акад. И.П. Павлова, Санкт-Петербург, Россия (журнал «Лучевая диагностика и терапия» №2, 2016) [читать];
статья «Мономелическая амиотрофия — редкий вариант болезни нижнего мотонейрона (2 клинических наблюдения)» Т.М. Алексеева, В.С. Демешонок, Н.Ю.Александров, А.Д. Халиков, М.Г. Соколова; ГБОУ ВПО Северо-Западный государственный медицинский университет имени И.И. Мечникова Минздрава России, Санкт-Петербург (журнал «Анналы клинической
и экспериментальной неврологии» №3, 2015) [читать];
статья «Предупрежден — значит вооружен: особенности МРТ-исследования при болезни Хираяма» Е.И. Кремнева, А.А. Воробьева, Л.С. Адарчева, Р.Н. Коновалов, А.С. Суслин, М.В. Кротенкова, М.Н. Захарова, Д.А. Гришина, А.Л. Антелава, В.В. Брюхов, А.В. Терехов; Научный центр неврологии, Москва, Россия; 1-я Республиканская клиническая больница Министерства здравоохранения Удмуртской Республики, г. Ижевск, Россия (журнал «Лучевая диагностика и терапия» №3, 2015) [читать];
статья (источник) «Болезнь Хираямы, или дистальная цервикальная миелопатия: новые варианты клинических и МРТ параметров» Е.Г.Менделевич, Л.К. Валиева, Э.И. Богданов (Казанский государственный медицинский университет, кафедра неврологии и реабилитации), В.И. Анисимов (РКБ МЗ РТ, отделение лучевой диагностики, Казань); журнал «Неврологический вестник» — 2015 — Т. XLVII, вып. 3 — С. 106 — 110 [читать];
статья «Болезнь Хираяма» на paindept.ru [читать];
статья «Клинический случай болезни Хираяма» Алексеева Т.М., Соколова М.Г., Демешонок В.С., Александров Н.Ю., Халиков А.Д.; СЗГМУ им. И.И. Мечникова, Санкт-Петербург; 2014 [читать];
статья «Синдром верхнего вялого парапареза при БАС и БАС-подобных синдромах: вопросы дифференциальной диагностики» М.Н. Захарова, И.В. Закройщикова, И.С. Бакулин, И.А. Кочергин; ФГБНУ Научный центр неврологии, Москва (журнал «Medica Mente» №1, 2016) [читать].
Источник
Зоопарк в голове
«Загляни глубже в природу, и ты поймешь все лучше.»
Альберт Эйнштейн
Изображения, получаемые при нейровизуализации, иногда вызывают ассоциации, например, с животными. Такие ассоциации легче запоминаются клиницистами в качестве маркеров различных нейродегенеративных заболеваний. Некоторые из них прошли испытание временем и стали общепризнанными. Другие ждут своей очереди быть описанными находчивыми врачами и стать представителями неврологического «зоопарка».
«Глаз тигра»
«Глаз тигра» впервые описан Sethi et al. в 1988 у двух пациентов с болезнью Галлервордена-Шпатца. Заболевание относится к группе наследственных нейродегенеративных заболеваний с аутосомно-рецессивным типом наследования. У большинства больных выявляется мутация гена пантотенаткиназы 2-го типа, приводящая к нарушению обмена железа. Поражаются преимущественно базальные ганглии головного мозга. При классической форме заболевание дебютирует в детском возрасте (средний возраст дебюта – 3 года) с дистонии конечностей или цервикальной дистонией с последующей генерализацией. Помимо дистонии, клиническими проявлениями могут быть спастические парезы, тремор, миоклония, хорея, эпиприпадки, паркинсонизм, когнитивные нарушения, пигментная ретинопатия и атрофия зрительного нерва. Через 5-10 лет пациенты теряют возможность к самостоятельному передвижению, появляются нарушения глотания и дыхания, что вскоре приводит к смерти. Клиника атипичных вариантов гетерогенна. При частичном снижении активности пантотенкиназы наблюдается дебют заболевания в подростковом или даже взрослом возрасте, течение заболевания более медленное. В клинической картине может доминировать паркинсонизм. Часто он сочетается оромандибулярной дистонией, дизартрией и нейропсихиатрическими нарушениями. Способность к самостоятельному передвижению утрачивается через 15-40 лет.
Формирование «глаза тигра» на изображениях МРТ головного мозга в Т2 режиме связано с поражением бледного шара. В центре отмечается повышение сигнала в связи с гибелью нейронов, глиозом. Вокруг формируется зона пониженного сигнала, что связано с патологическим накоплением железа. У небольшой части больных отмечается лишь снижение интенсивности сигнала от бледного шара. «Глаз тигра» считается патогномоничным признаком болезни Галлервордена-Шпатца, он присутствует более, чем в 95% случаев. Однако он может выявляться и при некоторых других заболеваниях с нарушением обмена железа и у клинически здоровых людей. (рисунок 1)
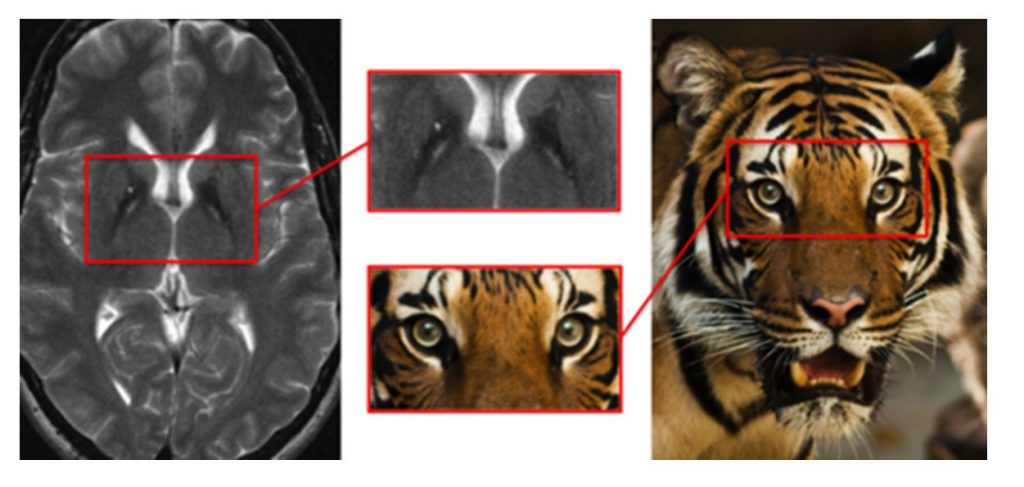
Рисунок 1. «Глаз тигра» при болезни Галлервордена-Шпатца.
«Мордочка панды»
Гепатолентикулярная дегенерация (болезнь Вильсона-Коновалова) – аутосомно-рецессивное заболевание, развивающееся в результате мутации в гене ATP7B. В основе заболевания лежит нарушение элиминации меди. Излишек меди накапливается в печени, головном мозге, роговице, почках и других органах. При дебюте в детском и подростковом возрасте болезнь чаще начинается с симптомов поражения печени, исходом которого может быть цирроз. В возрасте старше 20 лет болезнь чаще начинается с неврологических проявлений: тремора, дистонии, дизартрии, хореоатетоза, паркинсонизма, атаксии, аффективных и когнитивных нарушений. Гепатолентикулярную дегенерацию необходимо исключать у каждого больного с экстрапирамидным синдромом, проявившемся в возрасте до 50 лет.
В случае развития неврологической симптоматики при МРТ головного мозга всегда выявляются отклонения, такие как повышение сигнала в режиме Т2 от базальных ганглиев, таламуса, ствола и белого вещества полушарий. «Мордочку гигантской панды» можно увидеть на аксиальных срезах на уровне среднего мозга: гиперинтенсивный сигнал от покрышки, красных ядер и ретикулярной части черной субстанции, гипоинтенсивный сигнал от впроводящих путей. «Мордочку детеныша панды» можно увидеть на аксиальном срезе на уровне моста: гипоинтенсивный сигнал от покрышки и медиального продольного пучка («глаза»), верхних ножек мозжечка («ушки») и гиперинтенсивный сигнал вокруг водопровода («рот и нос») (рисунок 2).
Рисунок 2. «Мордочка панды» при болезни Вильсона-Коновалова.
«Хвост ласточки»
В патогенезе развития болезни Паркинсона большую роль играет поражение черной субстанции, особенно ее дорсолатеральной части. В норме при МРТ головного мозга в режиме ДВИ определяется гиперинтенсивность в каудальных и медиолатеральных отделах черной субстанции в виде «хвоста ласточки». К моменту клинического дебюта болезни Паркинсона наблюдается гибель 60-80% дофаминергических нейронов компактной части черной субстанции, что при нейровизуализации отражается как исчезновение «хвоста ласточки». Однако, данный симптом часто наблюдается и в случаях атипичного паркинсонизма и не позволяет проводить дифференциальную диагностику болезни Паркинсона и заболеваний, протекающих с синдромом паркинсонизма. (рисунок 3)
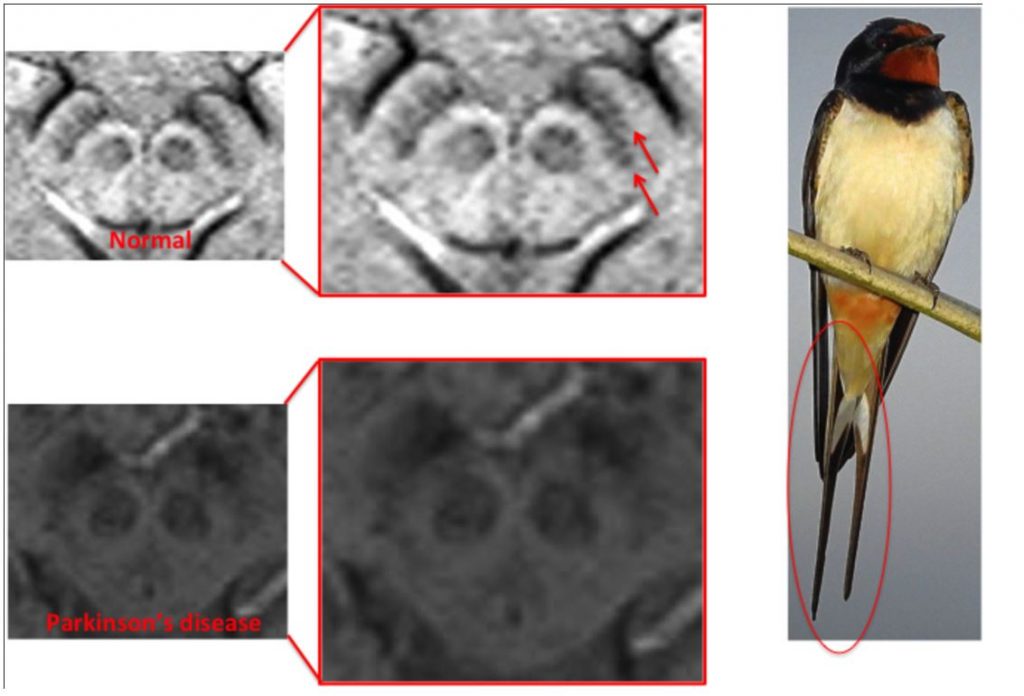
Рисунок 3. «Хвост ласточки» при паркинсонизме.
Симптомы колибри или королевского пингвина
и симптом Микки-Мауса.
Симптом колибри (или симптом королевского пингвина) можно выявить при нейровизуализации у пациентов с прогрессирующим надъядерным параличом. Клинически заболевание проявляется аксиальной ригидностью, псевдобульбарным синдромом, ранним развитием постуральной неустойчивости, когнитивным снижением надъядерным параличом вертикального взора. Симптом колибри отражает избирательную атрофию покрышки среднего мозга, затрагивающую ростральную часть интерстициального ядра медиального продольного пучка, с поражением которого связывают развитие паралича вертикального взора. При МРТ головного мозга на сагиттальном срезе видно пролабирование вниз крыши среднего мозга, что формирует очертания верхней поверхности головы и клюва «колибри» или «пингвина». Симптом может не выявляться на ранних стадиях заболевания, но является высокоспецифичным. Описаны случаи выявления «колибри» при нормотензивной гидроцефалии и сидроме ломкой Х-хромосомы. (рисунок 4)
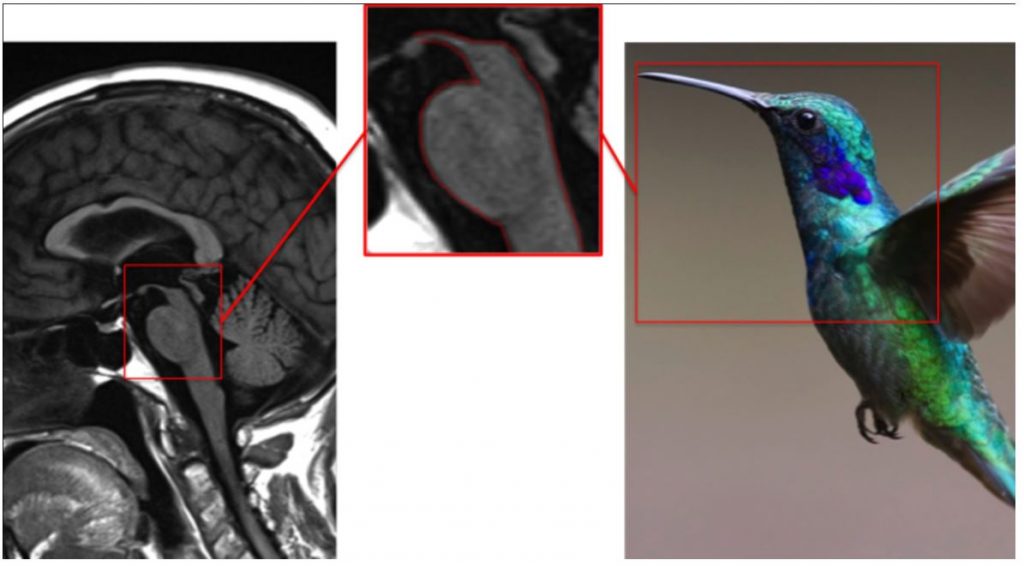
Рисунок 4. Симптом колибри при прогрессирующем надъядерном параличе.
Симптом Микки-Мауса можно выявить также при прогрессирующем надъядерном параличе, но уже на аксиальных срезах среднего мозга. Его атрофия приводит к «похуданию щечек мышонка». (рисунок 5)
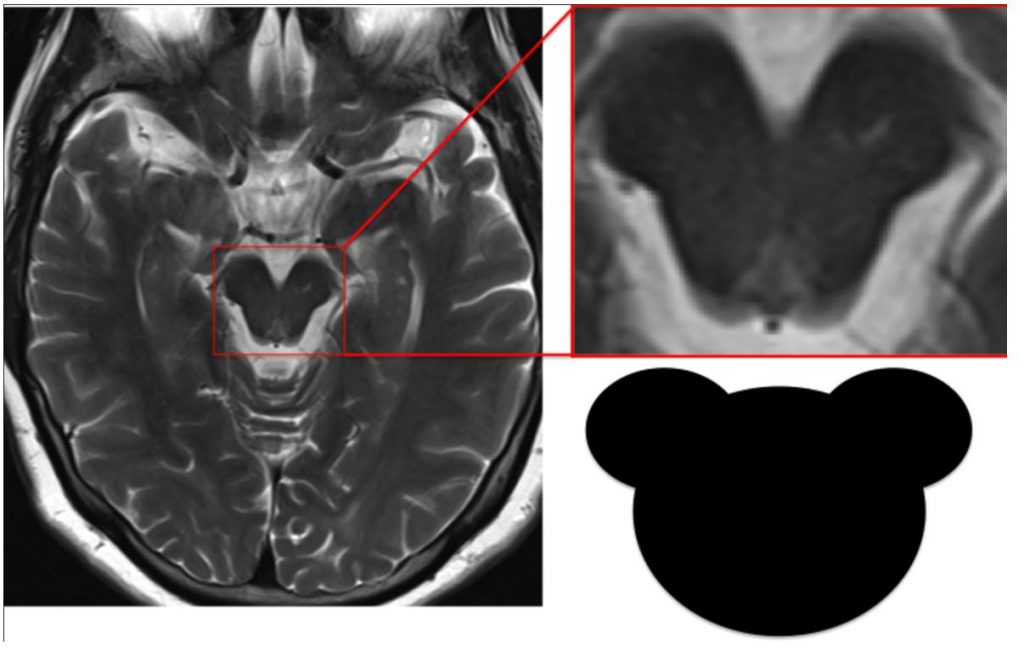
Рисунок 5. Симптом Микки-Мауса при прогрессирующем надъядерном параличе.
«Уши рыси»
Группа наследственных спастических параплегий включает большое количество заболеваний. Наследственные параплегии 11 и 15 типов клинически проявляются прогрессирующим спастическим нижним парапарезом в сочетании с когнитивными нарушениями и синдромом поражения двигательного нейрона. У части пациентов в дебюте развивается паркинсонизм или дистония. Начало заболевания как правило приходится на первое десятилетие жизни. При МРТ головного мозга выявляется истончение мозолистого тела. В режимах Т2 и FLAIR выявляется гиперинтенсивный сигнал от белого вещества около передних рогов боковых желудочков по форме напоминающий кисточки на ушах рыси. Похожие изменения могут выявляться при болезни Маркиафавы-Биньями, после коллозотомии или сочетаться с аномалиями развития мозолистого тела. (рисунок 6).
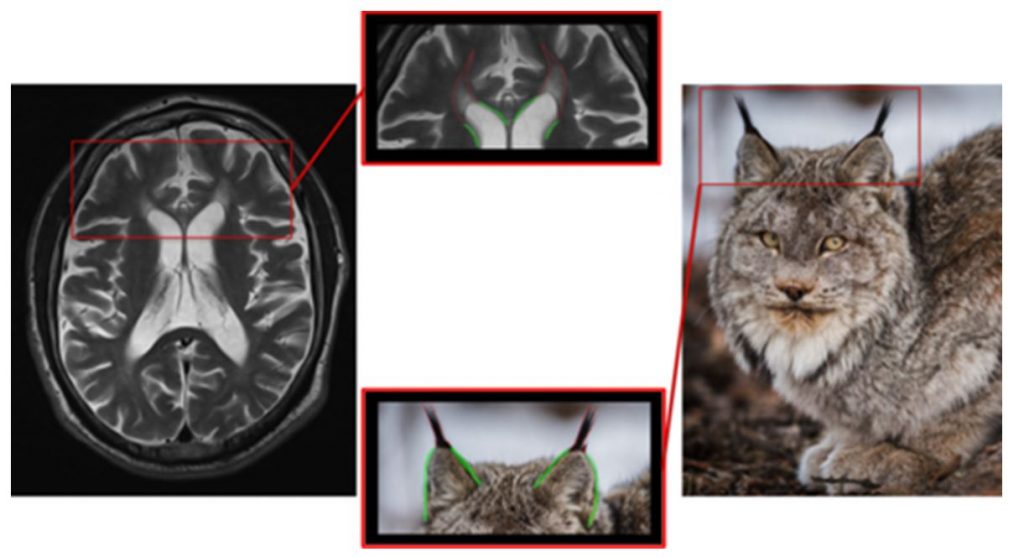
Рисунок 6. Уши рыси при наследственных спастических параплегиях.
Симптом стрекозы
Понтоцеребеллярная гипоплазия 2 типа – редкое аутосомно-рецессивное нейродегенеративное заболевание. Заболевание манифестирует в первые месяцы жизни – появляются трудности при кормлении, отставание в развитии, заторможенность. Типичны экстрапирамидные нарушения в виде хореоатетоза и дистонии. Пациентам с этим заболеванием часто ошибочно выставляется диагноз ДЦП. (рисунок 7)
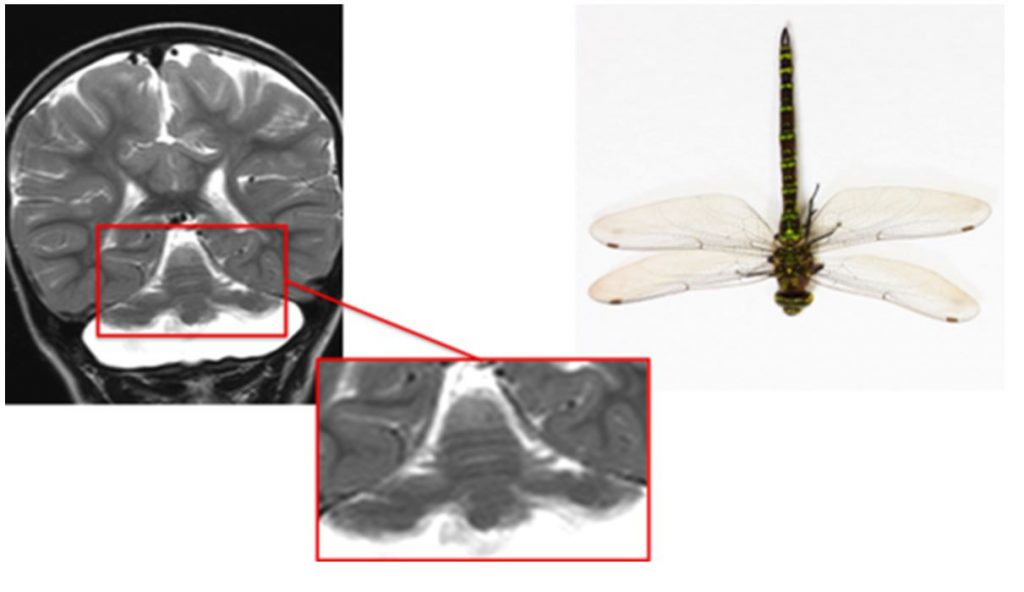
Симптом головастика
Болезнь Александера – наследственное демиелинизирующее заболевание с аутосомно-доминантным типом наследования. Классический вариант заболевания проявляется на первом году жизни, но возможен дебют и во взрослом возрасте. Клинически проявляется симптомами поражения ствола головного мозга, мозжечка и спинного мозга. Симптом головастика можно увидеть на сагиттальных срезах головного мозга вследствие выраженной атрофии продолговатого мозга и шейного отдела спинного мозга («хвостик головастика») при сохранности моста. Кроме симптома головастика, можно выявить изменение интенсивности сигнала от средних ножек мозжечка в режиме FLAIR. (рисунок 8)
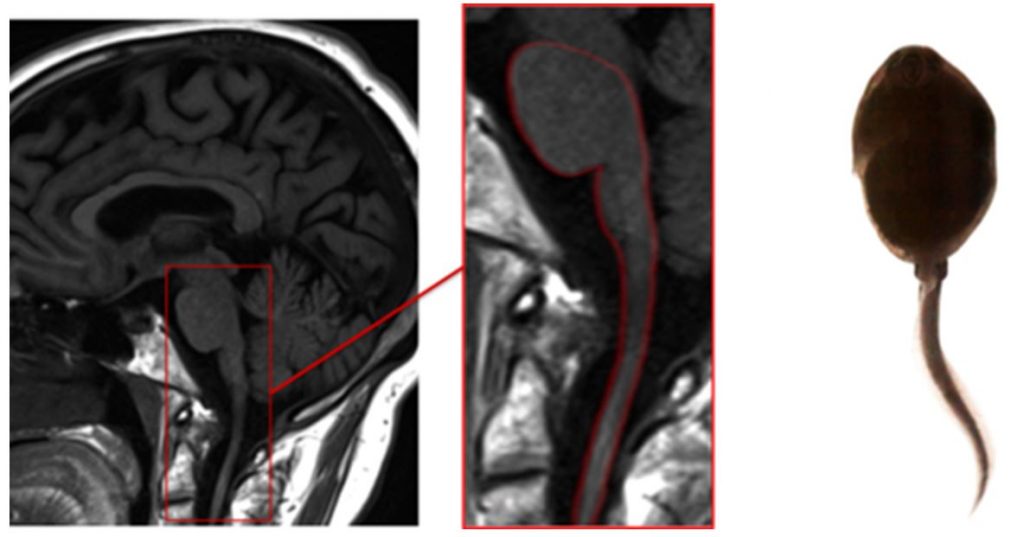
Рисунок 8. Симптом головастика при болезни Александера.
Симптом шкуры тигра или леопарда
Метахроматическая лейкодистрофия – наследственный липидоз с аутосомно-рецессивным типом наследования. В зависимости от возраста дебюта выделяют детскую, ювенильную и взрослую формы. Клинические проявления включают когнитивное снижение, атаксию и периферическую нейропатию. Изменения на МРТ головного мозга выявляются рано и коррелируют с выраженностью клинических проявлений. В режиме Т2 выявляется гиперинтенсивность мозолистого тела и перивентрикулярного белого вещества в лобной и теменной областях. Симптом шкуры тигра можно увидеть из-за чередования полос с нормальной интенсивностью сигнала и гиперинтенсивных. Феномен может наблюдаться и при других лизосомальных заболеваниях, при болезни Пелицеуса-Мерцбахера. (рисунок 9)
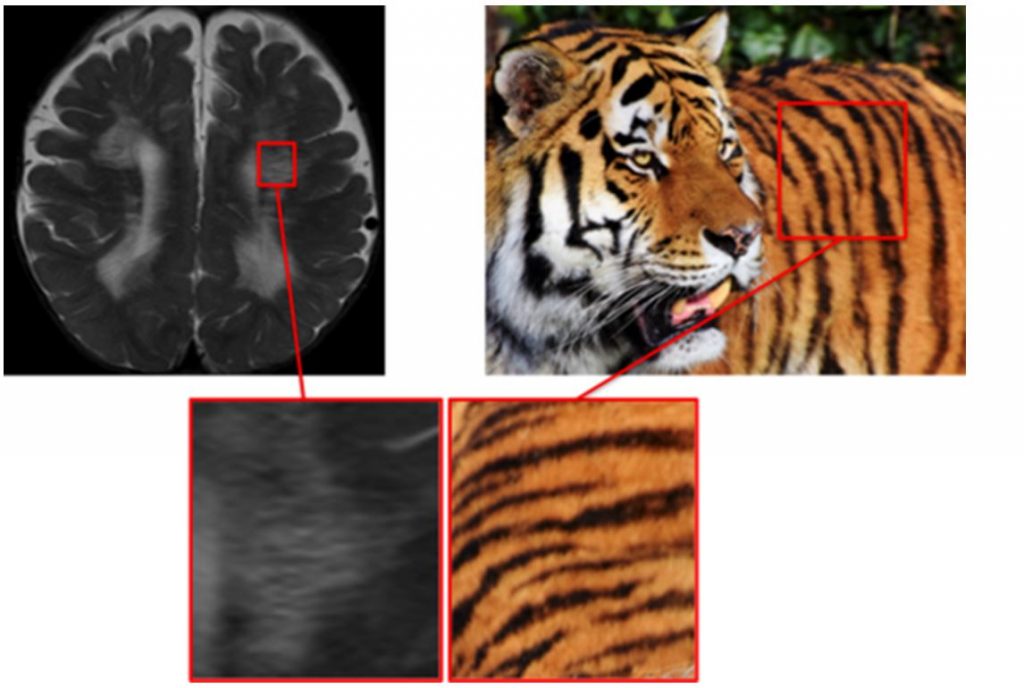
Рисунок 9. Симптом шкуры тигра при метахроматической лейкодистрофии.
Симптом крыла летучей мыши
Глутаровая ацидурия 1 типа – редкое аутосомно-рецессивное заболевание. Развивается вследствие нарушения метаболизма лизина и триптофана. Накопление токсичных продуктов метаболизма приводит к поражению стриатума с развитием дистонии. При МРТ головного мозга выявляется выраженная атрофия с значительным расширением Сильвиевой борозды в виде крыльев летучей мыши. Во многих странах на это заболевание проводится скрининг новорожденных. Строгая диета предотвращает развитие неврологической симптоматики.
Синдром Жубер – наследственное заболевание с манифестацией в неонатальном периоде. Клинически проявляется гипотонией, нарушениями дыхания, окуломоторной апраксией. Позднее присоединяются атаксия, когнитивные и двигательные нарушения. У пациентов, доживающих до взрослого возраста, могут развиться проявления паркинсонизма. При нейровизуализации выявляется аплазия червя мозжечка, удлинение верхних мозжечковых ножек в виде «симптома большого коренного зуба» и расширение четвертого желудочка в форме крыла летучей мыши. (рисунок 10)
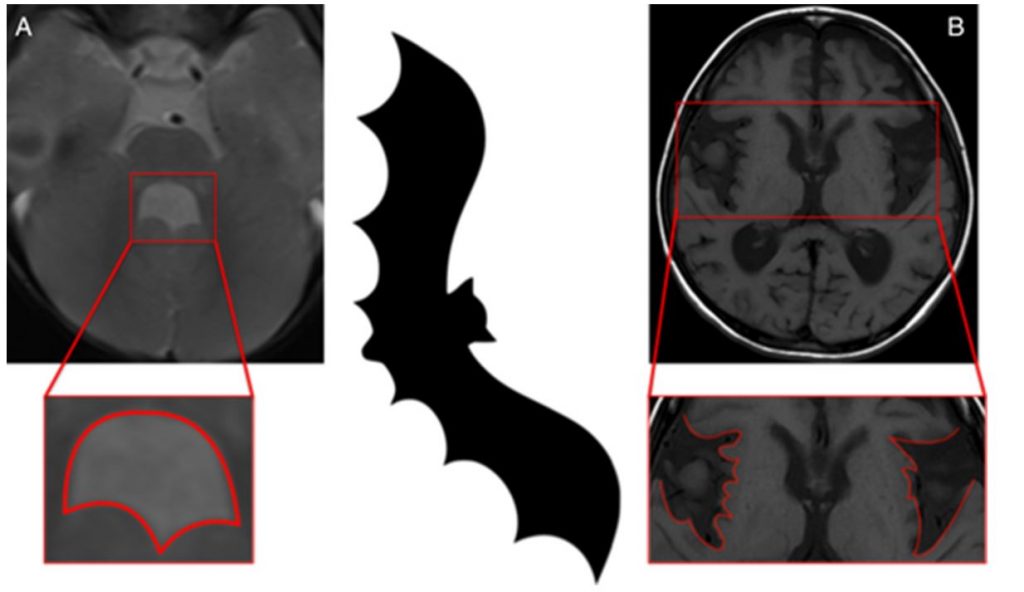
Рисунок 10. Четвертый желудочек в форме крыла летучей мыши при синдроме Жубер. Симптом крыльев летучей мыши при глутаровой ацидурии 1 типа.
Автор: Чеботарева А.Д.
Источник