Мрт головного мозга при болезни вильсона

Диагностика болезни Вильсона по КТ, МРТ головного мозгаа) Терминология: б) Визуализация: 1. Общие характеристики болезни Вильсона: 2. КТ признаки болезни Вильсона:
3. МРТ признаки болезни Вильсона: 4. Радионуклидная диагностика болезни Вильсона: 5. Рекомендации по визуализации: в) Дифференциалная диагностика: 1. Болезнь Лея: 2. Гипоксически-ишемическая энцефалопатия: 3. Болезнь Крейтцфельдта-Якоба: 4. Японский энцефалит (ЯЭ): 5. Органическая ацидурия: 6. Отравление метанолом: 7. Синдром осмотической демиелинизации:
г) Патология: 1. Общие характеристики болезни Вильсона: 2. Стадирование и классификация: 3. Макроскопические и хирургические особенности: 4. Микроскопия:
д) Клиническая картина: 1. Проявления болезни Вильсона: 2. Демография: 3. Течение и прогноз: 4. Лечение: е) Диагностическая памятка. Советы по интерпретации изображений: ж) Список литературы:
— Также рекомендуем «Диагностика токсического и метаболического поражения головного мозга» Редактор: Искандер Милевски. Дата публикации: 26.4.2019 |
Источник
Согласно литературным данным, нет симптомов, специфичных для этого заболевания, нет типичной клинической картины, что является причиной запоздалой диагностики этой патологии. И.К. Волошин-Гапонов в своей статье цитирует слова J. Walshe: «Нет двух одинаковых пациентов, даже если они кровные родственники и нет такой вещи, как типичная клиника болезни Вильсона».
Болезнь Вильсона — Коновалова (БВК, гепато-лентикулярная дегенерация, гепато-церебральная дегенерация) — тяжелое заболевание, обусловленное генетически детерминированным нарушением обмена меди. Заболевание возникает с частотой 1 — 9 : 100000 населения (в среднем 1 на 25000), может проявиться в большом возрастном диапазоне, от 5 до 50 и более лет.
Патогенез. Ответственный ген АТР7В расположен в длинной части 13-й хромосомы (участок 13q14q21) и передается по отцовской линии (наследуется по аутосомно-рецессивному типу; носителем дефектного гена по ориентировочным оценкам является каждый 90 — 100-ый человек [1%]; вероятность рождения больного ребенка у родителей-гетерозигот составляет 25%). Ген ATP7B экспрессируется, в основном, в печени и кодирует медь-транспортирующую АТФ-азу 7В (син.: АТФ-аза 2, АТФ-аза Р-типа). АТФ-аза 7В имеет 6 медь-связывающих участков. Она осуществляет транспорт ионов меди из клетки за счет энергии расщепления АТФ, а также участвует в образовании из апо-церулоплазмина функционально активного церулоплазмина (процесс «нагрузки» апо-церулоплазмина медью с образованием функционально активного церулоплазмина), который затем выделяется в кровь (церулоплазмин относится к альфа2-глобулинам и синтезируется исключительно в печени, в цитоплазме гепатоцитов вокруг ядра). Отсутствие АТФ-азы 7В нарушает выделение меди из головного мозга в кровь, из крови в желчь (билиарная экскреция меди) и далее с калом из организма. Таким образом, при БКВ свободная медь не элиминируется, а откладываясь в различных органах обусловливает многообразие симптомов. Неврологические проявления болезни определяются накоплением меди в структурах головного мозга и нарушением функции печени.
Ведущим звеном патогенеза БВК является хроническая интоксикация медью. Медь накапливается в печени, селезенке, почках, головном мозге, роговице, хрусталике глаза и других органах. Накопление меди в печени приводит к некрозу гепатоцитов, воспалению, фиброзу, пролиферации желчных протоков и циррозу; в головном мозге — к некрозу нейронов с образованием полостей (кист). Изменения других органов и тканей, как правило, незначительны.
С пищей за сутки в организм поступает 2 — 5 мг меди. Она всасывается в кишечнике, поступает в печень, где связывается с синтезируемым печенью церулоплазмином, циркулирует в сыворотке крови, избирательно захватывается органами, которые в ней нуждаются, а экскретируется с желчью. Незначительная часть меди находится в крови в ионизированной форме в виде лабильного комплекса с альбумином и выделяется с мочой. Снижение или отсутствие активности церулоплазмина нарушает поступление достаточных количеств меди к ферментам тканевого дыхания, кроветворным органам; свободная медь, накапливающаяся в тканях, блокирует SH-группы многих ферментов. Следствием недостаточного использования меди является ее депонирование в печени, мозге, почках, роговице. Складывается парадоксальная ситуация: нарушение биологических процессов из-за недостаточного количества меди и накопление меди в тканях с симптоматикой интоксикации металлом.
Клиника. Ведущими в клинике БВК являются различные экстрапирамидные расстройства, мозжечковые и психические нарушения. В итоге из их сочетания возникает сложная симфония симптомов, диагностика представляет значительные трудности, опорными моментами являются в основном результаты лабораторных исследований и наличие кольца Кайзера — Флешнера (см. далее).
У молодых, в возрасте до 20 лет болезнь чаще начинается с симптомов поражения печени, исходом может быть цирроз печени. Реже поражаются почки, поджелудочная железа, сердце. В последующем присоединяются неврологические симптомы. В отсутствие лечения летальный исход наступает в течение нескольких лет. В возрасте более 20 лет дебют гепато-лентикулярной дегенерации чаще с развития неврологических расстройств: тремор, дистония, хорея, атаксия, акинезия, дизартрия и протекает медленнее (тремор возникает в 50% случаев, со временем гиперкинез становится более распространенным и грубым, напоминая «порхающие крылья»).
Для большинства форм характерно развитие ригидности мышц, которая может быть в числе первых симптомов, и в случае вовлечения ног вынуждает больных ходить на носках. Рано меняется, обедняясь, мимика, в последующем появляются гримасы насильственного плача или смеха. Почти облигатным симптомом является нарушение речи, имеет место своеобразная форма моторной афазии — подкорковая, при которой моторный речевой центр Брока не является ведущим. У 10 — 20% больных в качестве первых симптомов возникают психические нарушения.
Существует несколько классификаций, основанных на клинических проявлениях БВК. Н.В. Коновалов выделяет 5 форм заболевания:
[1] ригидно-аритмогиперкинетическую (раннюю);
[2] дрожательную (позднюю);
[3] дрожательно-ригидную;
[4] экстрапирамидно-корковую;
[5] абдоминальную.
Таким образом, основными формами БВК являются абдоминальная [5] и церебральная [1, 2, 3, 4]. Абдоминальная форма может проявляться гепатопатией, вильсоновским гепатитом и циррозом печени, фульминантной печеночной несостоятельностью.
По данным молекулярно-генетических исследований некоторые авторы выделяют 3 генетических типа БВК:
[1] славянский — с поздним дебютом неврологических симптомов в 20 — 35лет и минимальным вовлечением печени;
[2] западный тип — дебютирует в 10 — 16 лет поражением печени с более поздними неврологическими проявлениями, встречается в Европе, Средней Азии и Китае;
[3] атипический — без клинических проявлений, с изолированным снижением уровня церулоплазмина.
Течение заболевания можно разделить на 2 стадии: [1] латентную и [2] стадию клинических проявлений. При латентной стадии клинические симптомы отсутствуют и признаки заболевания (признаки накопления меди в организме) выявляются только при лабораторном обследовании. При эффективном лечении дополнительно выделяют [!] стадию отрицательного баланса меди. В этом случае в результате достижения и длительного поддержания отрицательного баланса меди наблюдается регресс клинических и лабораторных проявлений заболевания.
Диагностика БВК базируется на комбинации клинических симптомов, данных лабораторного обследования и молекулярно-генетического тестирования и включает в себя следующие параметры:
■ снижение уровня церулоплазмина в сыворотке крови (менее 20 мг/дл);
■ повышение суточной экскреции меди с мочой: базальной — более 50 мкг/сут, в пробе с Д-пенициллмином (500 мг х 2 раза /сут) — более 1600 мкг/сут;
■ повышение количественного содержания меди в ткани печени более 250 мкг/г (или положительная гистохимическая окраска на медь печеночной ткани);
■ наличие колец Кайзера — Флейшера [см. фото. справа] (медная катаракта по типу «подсолнух») при офтальмологическом осмотре (обнаруживается в 95% случаях при неврологических проявлениях БВК);
■ наличие специфической нейропсихиатрической симптоматики (или изменения на МРТ головного мозга);
■ гомозиготное/компаундгетерозиготное носительство мутаций гена АТР 7В.
Ведущими в диагностике ГЦД являются лабораторные данные и наличие роговичного кольца Кайзера-Флейшнера. В сомнительных случаях прибегают к биопсии печени или радиоизотопному исследованию.
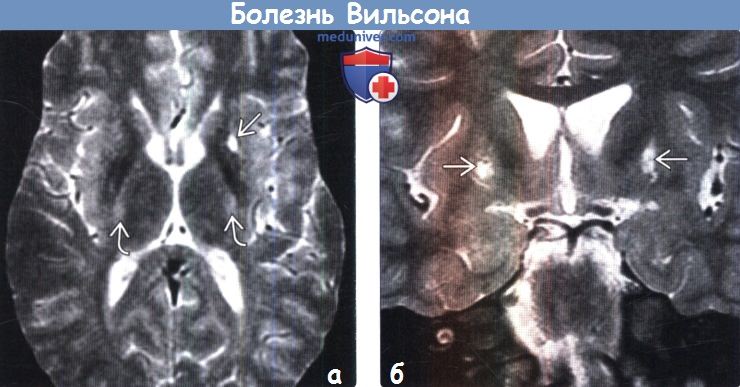
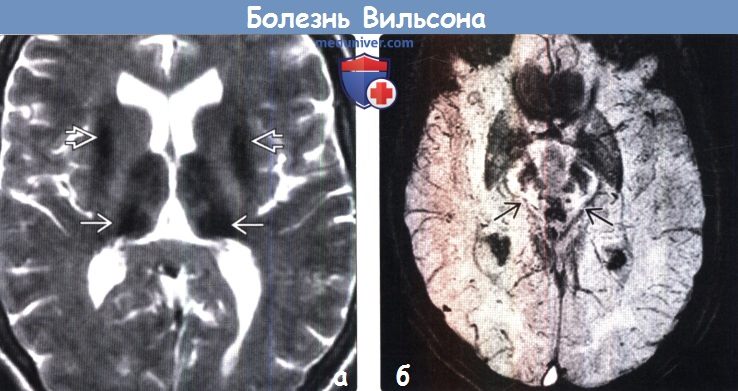
Для выявления характерных структурных изменений в ЦНС при БВК целесообразно использовать магнитно-резонансную томографию (МРТ) или компьютерную томографию (КТ) головного мозга. Расширение желудочков, атрофия коры и ствола мозга при КТ головного мозга наблюдаются чаще, чем билатеральные зоны пониженной плотности в области базальных ганглиев. Более важным диагностическим методом при церебральной форме заболевания является МРТ. Патология головного мозга на МРТ обнаруживается у всех пациентов с БВК. МРТ выявляет характерные очаги в головном мозге, а также неспецифическую диффузную атрофию мозга. Типична гиперинтенсивность сигнала в Т2-режиме в области чечевицеобразных, таламических и хвостатых ядер, ствола мозга, мозжечка и белого вещества. Характерным, но редким МРТ-симптомом в срезах среднего мозга является картина, напоминающая «лицо гигантской панды» (см. МРТ-скан слева). КТ- и МРТ-симптомы в ряде случаев могут опережать появление клинических симптомов.
читайте также статью «Структурные изменения головного мозга у больных с гепатоцеребральной дегенерацией» Волошин-Гапонов И.К., ГУ «Институт неврологии, психиатрии и наркологии НАМН Украины», г. Харьков (Международный неврологический журнал 2 (56), 2013) [читать]
Прогноз БВК определяется сроком начала терапии (при своевременной диагностике БВК успешно поддается терапии). Если лечение начато до появления неврологических симптомов, то последние не возникают, а продолжительность жизни не уменьшается. Лечение, начатое после появления неврологических симптомов лишь в 20% дает полную ремиссию, и в 60 — 70% ремиссию частичную.
В настоящее время используются различные препараты для лечения БВК, обладающие различным строением и механизмом действия. Д-пеницилламин является препаратом выбора при лечении БВК. Д- пеницилламин представляет собой производное пенициллина, молекула которого содержит свободную сульфидрильную группу, с помощью которой осуществляется хелаторная активность данного препарата. Д-пеницилламин мобилизует медь из печени и других органов и увеличивает ее мочевую экскрецию, а также индуцирует синтез 19 металлотионинов. Д-пеницилламин назначается за 1 час или через 2 часа после приема пищи, т.к. еда снижает кишечную абсорбцию препарата. Начальные дозы при терапии составляют 250-500 мг/сут с постепенным (каждые 4 — 7 дней) увеличением дозы на 250 мг до лечебной дозировки 1000 — 1500 мг/сут, которая дается в 2 — 4 приема. Для детей терапевтическая дозировка Д-пеницилламина составляет 20 мг/кг/сут. Коррекция дозы препарата производится по мере снижения уровня экскреции меди с мочой. При применении Д-пеницилламина развивается недостаточность пиридоксина, что требует назначения витамина В6 (пиридоксин) в дозе 25-50 мг/сут. При развитии побочных действий Д-пеницилламина назначается другое альтернативное лечение: препараты цинка (сульфат, окись, ацетат цинка), триентин (триэтилен-тетрамин), тетрамолибдат аммония (последние два препарата на Российском фармакологическом рынке не зарегистрированы), трансплантация печени.
читайте также статью «Препараты, противопоказанные при миастении. Клинические наблюдения манифестации скрытых форм миастении после применения D-пеницилламина, тимолола, сульфата магния, ботулинического токсина типа А, мерказолила» Щербакова Н.И., Хрущева Н.А., Супонева Н.А., Антонова К.В., Белова Н.В., Шведков В.В., Шабалина А.А., Костырева М.В., Галкина О.И., Рудниченко В.А., Касаткина Л.Ф.; ФГБНУ «Научный центр неврологии», Москва; ФГБОУ ВПО «Первый МГМУ им. И.М. Сеченова» МЗ РФ, Москва (Неврологический журнал, №3, 2017) [читать]
Дополнительная информация:
[1] Федеральные клинические рекомендации по диагностике и лечению болезни Вильсона-Коновалова (гепатолентикулярная дегенерация), МЗ РФ, 2013 [читать]
[2] Федеральные клинические рекомендации по оказанию медицинской помощи детям с болезнью Вильсона (МЗ РФ, Союз педиатров России), 2015 [читать]
статья «Болезнь Вильсона — Коновалова: «великий хамелеон» Пономарев В.В., 5-я клиническая больница, г. Минск, Республика Беларусь (Международный неврологический журнал, №3 (33), 2010) [читать]
статья «Клиника церебральных нарушений при гепатоцеребральной дегенерации» Волошин-Гапонов И.К., ГУ «Институт неврологии, психиатрии и наркологии НАМН Украины» (Международный неврологический журнал, №2 (64), 2014) [читать]
статья «Современные аспекты болезни Вильсона» Мищенко Т.С., Волошин-Гапонов И.К., ГУ «Институт неврологии, психиатрии и наркологии НАМН Украины», г. Харьков (Международный неврологический журнал, № 2 (72), 2015) [читать]
Источник